Welcome to coolpup.py’s documentation!¶
Project homepage with code and issues is on GitHub.
Please feel free to report any problems or contribute.
coolpup.py¶
![]()
![]()
![]()
![]()
.cool file pile-ups with python.
A versatile tool to perform pile-up analysis on Hi-C data in .cool format (https://github.com/mirnylab/cooler). And who doesn’t like cool pupppies?
Introduction¶
What are pileups?¶
Pileups is the generic term we use to describe any procedure that averages multiple 2D regions (snippets) of a 2D matrix, e.g. Hi-C data. In some contexts they are also known as APA (aggregate peak analysis, from Rao et al., 2014), or aggregate region/TAD analysis (in GENOVA, van der Weide et al., 2021), and other names. The most typical use case is to quantify average strength of called dots (loops) in Hi-C data, or strength of TAD boundaries. However the approach can do much more than that. This is the idea of how pileups work to check whether certain regions tend to interact with each other:
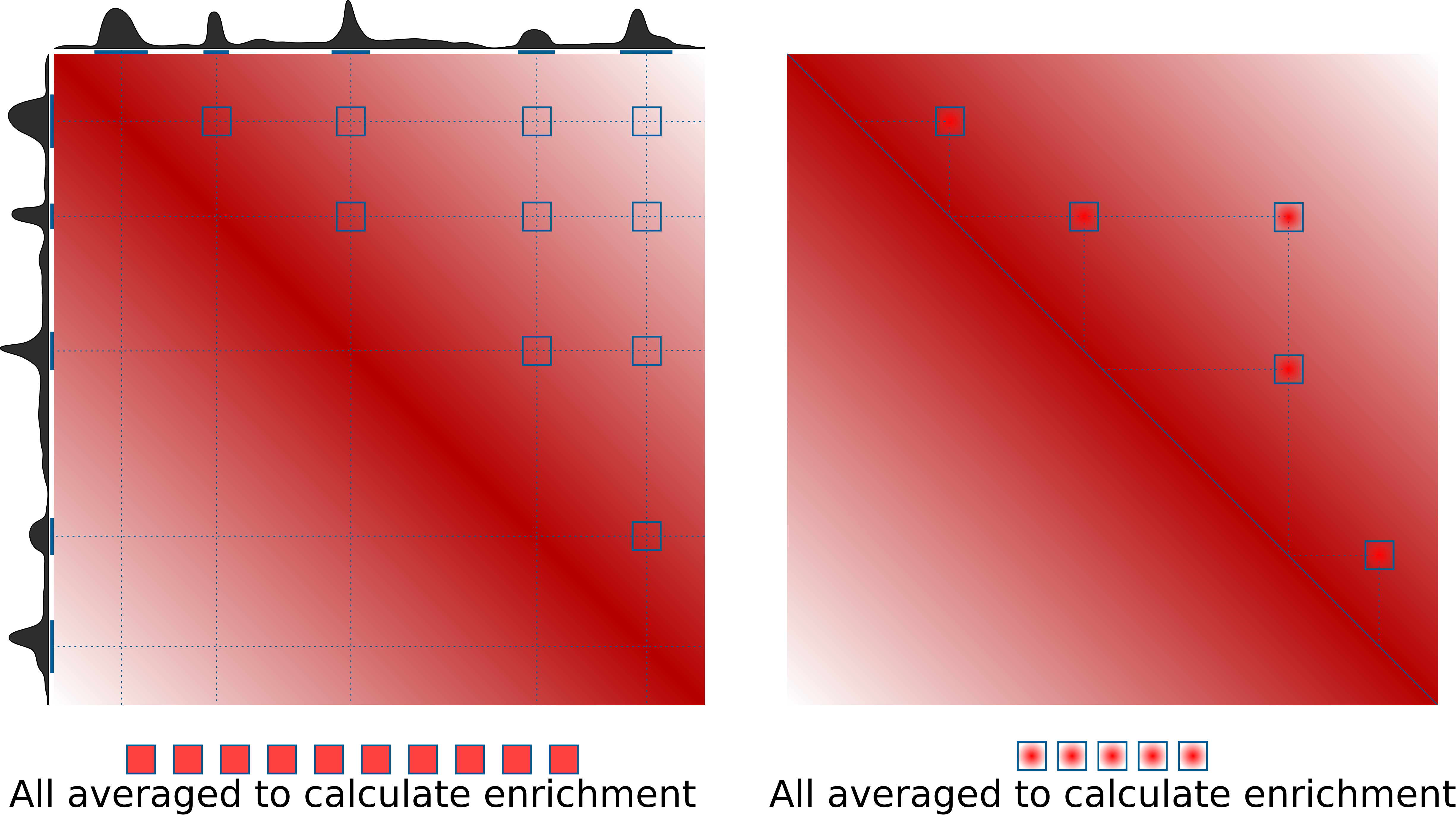
On the right is the more typical use case for quantification of loop strength. On the left is a different approach, designed to check whether specific regions in the genome (e.g. binding sites of a certain factor) tend to interact with each other.
What is very important for this quantification, is the normalization to expected values. This can be done in two ways: either using a chromosome- (or arm-) wide by-distance expected interactions, using a file with average values of interactions at different distances (e.g. output of cooltools expected-cis), or directly from Hi-C data by dividing the pileups over randomly shifted control regions. If neither expected normalization approach is used (just set --nshifts 0), this becomes essentially identical to the APA approach (Rao et al., 2014), which can be used for averaging strongly interacting regions, e.g. annotated loops. For weaker interactors, decay of contact probability with distance can hide any focal enrichment that could be observed otherwise. However, most importantly, when comparing different sets of regions at even slightly different distances, or comparing different datasets, the decay of contact probability with distance will very strongly affect the resulting values, hence normalizing to it is essential in many cases, and generally recommended.
coolpup.py vs cooltools pileup¶
cooltools is the main package with Hi-C analysis maintained by open2C. It also has a tool to perform pileups. Why does coolpup.py exit then?
The way cooltools pileup works, is it accumulates all snippets for the pileup into one 3D array (stack). Which gives a lot of flexibility in case one wants to subset the snippets based on some features later, or do some other non-standard computations based on the stack. But this is only advantageous when one performs analysis using the Python API, and moreover limits the application of cooltools pileup so it can’t be applied to a truly large number of snippets due to memory requirements. That’s where coolpup.py comes in: internally it never stores more than one snippet in memory, hence there is no limit to how many snippets can be processed. coolpup.py is particularly well suited performance-wise for analysing huge numbers of potential interactions, since it loads whole chromosomes into memory one by one (or in parallel to speed it up) to extract small submatrices quickly. Having to read everything into memory makes it relatively slow for small numbers of loops, but performance doesn’t decrease until you reach a huge number of interactions. Additionally, cooltools pileup doesn’t support inter-chromosomal (trans) pileups, however it is possible in coolpup.py.
While there is no way to subset the snippets after the pileup is generated (since they are not stored), coolpup.py allows one to perform various subsetting during the pileup procedure. Builtin options in the CLI are subsetting by distance, by strand, by strand and distance at the same time, and by window/region - in case of a provided BED file, one pileup is generated for each row against all others in the same chromosome; in case of trans-pileups, pileups for each chromosome pair can be generated. Importantly, in Python API any arbitrary grouping of snippets is possible.
.cool format¶
.cool is a modern and flexible format to store Hi-C data.
It uses HDF5 to store a sparse representation of the Hi-C data, which allows low memory requirements when dealing with high resolution datasets. Another popular format to store Hi-C data, .hic, can be converted into .cool files using hic2cool (https://github.com/4dn-dcic/hic2cool).
See for details:
Abdennur, N., and Mirny, L. (2019). Cooler: scalable storage for Hi-C data and other genomically-labeled arrays. Bioinformatics. 10.1093/bioinformatics/btz540
Getting started¶
Installation¶
All requirements apart are available from PyPI or conda.
Before installing everything you need to obtain Cython using either pip or conda. Then for coolpuppy (and other dependencies) simply do:
pip install coolpuppy
or
pip install https://github.com/open2c/coolpuppy/archive/master.zip
to get the latest version from GitHub. This will make coolpup.py callable in your terminal, and importable in python as coolpuppy.
Usage¶
The basic usage syntax is as follows:
coolpup.py [OPTIONS] coolfile.cool regionfile.bed
A guide walkthrough to pile-up analysis is available here (WIP): Walkthrough
Docs for the command line interface are available here: CLI docs
Some examples to get you started with CLI interface are available here and for the python API examples see here.
Plotting results¶
For flexible plotting, I suggest to use matplotlib or another library. However simple plotting capabilities are included in this package. Just run plotpup.py with desired options and list all the output files of coolpup.py you’d like to plot.
Citing coolpup.py¶
Ilya M Flyamer, Robert S Illingworth, Wendy A Bickmore (2020). Coolpup.py: versatile pile-up analysis of Hi-C data. Bioinformatics, 36, 10, 2980–2985.
https://academic.oup.com/bioinformatics/article/36/10/2980/5719023
doi: 10.1093/bioinformatics/btaa073
Guide to pileup analysis¶
Coolpup.py is a tool for pileup analysis. But what are pile-ups?
If you don’t know, you might have seen average ChIP-seq or ATAC-seq profiles which look something like this:

Pile-ups in Hi-C are essentially the same as average profiles, but in 2 dimensions, since Hi-C data is a a matrix, not a linear track!
Therefore instead of a linear plot, pileups are usually represented as heatmaps - by mapping values of different pixels in the average matrix to specific colours.
Pile-ups of interactions between a set of regions¶
For example, we can again start with ChIP-seq peaks, but instead of averaging ChIP-seq data around them, combine them with Hi-C data and check whether these regions are often found in proximity to each other. The algorithm is simple: we find intersections of all peaks in the Hi-C matrix (with some padding around the peak), and average them. If the peaks often interact, we will detect an enrichment in the centre of the average matrix:

Here is a real example:

Here I averaged all (intra-chromosomal) interactions between highly enriched ChIP-seq peaks of RING1B in mouse ES cells. I added 100 kbp padding to each bin containing the peak, and since I used 5 kbp resolution Hi-C data, the total length of each side of this heatmap is 205 kbp. I also normalizes the result by what we would expect to find by chance, and therefore the values indicate observed/expected enrichment. Because of that, the colour is log-scaled, so that the neutral grey colour corresponds to 1 - no enrichment or depletion, while red and blue correspond to value above and below 1, respectively.
What is important, is that in the center we see higher values than on the edges: this means that regions bound by RING1B tend to stick together more, than expected! The actual value in the central pixel is displayed on top left for reference.
This analysis is the default mode when coolpup.py is run with a .bed file, e.g. coolpup.py my_hic_data.cool my_protein_peaks.bed (with optional --expected my_hic_data_expected.tsv for normalization to the background level of interactions).
Pile-ups of predefined regions pairs, e.g. loops¶
A similar approach is based on averaging predefined 2D regions corresponding to interactions of specific pairs of regions. A typical example would be averaging loop annotations. This is very useful to quantify global perturbations of loop strength (e.g. annotate loops in WT, compare their strength in WT vs KO of an architectural protein), or to quantify them in data that are too sparse, such as single-cell Hi-C. The algorithm is very simple:

And here is a real example of CTCF-associated loops in ES cells:

Comparing with the previous example, you can clearly see that if you average loops that have been previously identified you, of course, get much higher enrichment of interactions, than if you are looking for a tendency of some regions to interact.
This analysis is performed with coolpup.py when instead of a bed file you provide a .bedpe file, so simply coolpup.py my_hic_data.cool my_loops.bedpe (with optional --expected my_hic_data_expected.tsv for normalization to the background level of interactions). bedpe is a simple tab-separated 6-column file with chrom1, start1, end1, chrom2, start2, end2.
Local pileups¶
A very similar approach can be used to quantify local properties in Hi-C maps, such as insulation. Valleys of insulation score can be identified (e.g. using cooltools diamond-insulation), or another way of identifying potential TAD boundaries can be used. Then regions around their positions can be averaged, and this can be used to visualize and quantify insulation in the same or another sample:

Here is an example of averaged insulation score valleys in mouse ES cells:

One can easily observe that these regions on average indeed have some insulating properties, and moreover the stripes emanating from the boundaries are very clear - they are a combination of real stripes found on edges of TADs in many cases, and loops found at different distances from the boundary in different TADs.
Average insulation can be quantiifed by dividing signal in two red squares (top left and bottom right corners) by the signal in the more blue squares (top right and bottom left corners), and here it is shown in the top left corner.
This analysis is very easily performed using coolpup.py: simply run coolpup.py my_hic_data.cool my_insulating_regions.bed --local (with optional --expected my_hic_data_expected.tsv for normalization to the background level of interactions; note that for local analyses in my experience random shift controls work better).
Rescaled pileups¶
If instead of boundary regions you have, for example, annotation of long domains, such as TADs, you can also average them to analyse internal interactions within these domains. The problem with simply applying the previous analysis to this case is that these domains can be of different length, and direct averaging will produce nonsensical results. However the submatrices corresponding to interactions within each domain (with some padding around) can all be individually rescaled to the same size, and then averaged. This way boundaries of these domains would be aligned in the final averaged matrices, and the pileups would make sense! here is a visual explanation of this idea:

And here is an example of such local rescaled pileups of TADs annotated using insulation score valleys used above in ES cells:

Each TAD was padded with the flanks of the same length as the TAD, and then they were all rescaled to 99x99 pixels. The pattern of the average TAD is very clear, in particular the corner loop at the top of the domain is highly enriched. Also the stripes, indicative of loop extrusion, both on the TAD borders and outside the TADs, are clearly visible.
You might notice that I removed the few central diagonals of the matrix here. That is because they are often noisy after this rescaling procedure, depending on the data you use.
To perform this analysis, you simply need to call coolpup.py my_hic_data.cool my_domains.bed --rescale --local (with optional --expected my_hic_data_expected.tsv for normalization to the background level of interactions). To specify the side of the final matrix as 99 bins add --rescale_size 99. Another useful option here is --rescale_pad, which defines the fraction of the original regions to use when padding them; the default value is 1, so each TAD is flanked on each side by a region of the same size.
Coolpuppy python API walkthrough notebook¶
Please see https://github.com/open2c/open2c_examples for detailed explanation of how snipping and pileups work, and explanation of some terminology
# If you are a developer, you may want to reload the packages on a fly.
# Jupyter has a magic for this particular purpose:
%load_ext autoreload
%autoreload 2
# import standard python libraries
import matplotlib as mpl
%matplotlib inline
mpl.rcParams['figure.dpi'] = 96
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
# import libraries for biological data analysis
from coolpuppy import coolpup
from coolpuppy.lib import numutils
from coolpuppy.lib.puputils import divide_pups
from coolpuppy import plotpup
import cooler
import bioframe
import cooltools
from cooltools import expected_cis, expected_trans
from cooltools.lib import plotting
Download data¶
For the test, we collected the data from immortalized human foreskin fibroblast cell line HFFc6:
Micro-C data from Krietenstein et al. 2020
ChIP-Seq for CTCF from ENCODE ENCSR000DWQ
You can automatically download test datasets with cooltools. More information on the files and how they were obtained is available from the datasets description.
# Print available datasets for download
cooltools.print_available_datasets()
1) HFF_MicroC : Micro-C data from HFF human cells for two chromosomes (hg38) in a multi-resolution mcool format.
Downloaded from https://osf.io/3h9js/download
Stored as test.mcool
Original md5sum: e4a0fc25c8dc3d38e9065fd74c565dd1
2) HFF_CTCF_fc : ChIP-Seq fold change over input with CTCF antibodies in HFF cells (hg38). Downloaded from ENCODE ENCSR000DWQ, ENCFF761RHS.bigWig file
Downloaded from https://osf.io/w92u3/download
Stored as test_CTCF.bigWig
Original md5sum: 62429de974b5b4a379578cc85adc65a3
3) HFF_CTCF_binding : Binding sites called from CTCF ChIP-Seq peaks for HFF cells (hg38). Peaks are from ENCODE ENCSR000DWQ, ENCFF498QCT.bed file. The motifs are called with gimmemotifs (options --nreport 1 --cutoff 0), with JASPAR pwm MA0139.
Downloaded from https://osf.io/c9pwe/download
Stored as test_CTCF.bed.gz
Original md5sum: 61ecfdfa821571a8e0ea362e8fd48f63
# Downloading test data for pileups
# cache = True will download the data only if it was not previously downloaded
# data_dir="./" will force download to the current directory
cool_file = cooltools.download_data("HFF_MicroC", cache=True, data_dir='./')
ctcf_peaks_file = cooltools.download_data("HFF_CTCF_binding", cache=True, data_dir='./')
ctcf_fc_file = cooltools.download_data("HFF_CTCF_fc", cache=True, data_dir='./')
resolution = 10000
# Open cool file with Micro-C data:
clr = cooler.Cooler(f'{cool_file}::/resolutions/{resolution}')
# Use bioframe to fetch the genomic features from the UCSC.
hg38_chromsizes = bioframe.fetch_chromsizes('hg38')
hg38_cens = bioframe.fetch_centromeres('hg38')
hg38_arms = bioframe.make_chromarms(hg38_chromsizes, hg38_cens)
# Select only chromosomes that are present in the cooler.
# This step is typically not required! we call it only because the test data are reduced.
hg38_arms = hg38_arms.set_index("chrom").loc[clr.chromnames].reset_index()
# call this to automaticly assign names to chromosomal arms:
hg38_arms = bioframe.make_viewframe(hg38_arms)
hg38_arms
| chrom | start | end | name | |
|---|---|---|---|---|
| 0 | chr2 | 0 | 93139351 | chr2_p |
| 1 | chr2 | 93139351 | 242193529 | chr2_q |
| 2 | chr17 | 0 | 24714921 | chr17_p |
| 3 | chr17 | 24714921 | 83257441 | chr17_q |
hg38_arms.to_csv('hg38_arms.bed', sep='\t', header=False, index=False) # To use in CLI
# Read CTCF peaks data and select only chromosomes present in cooler:
ctcf = bioframe.read_table(ctcf_peaks_file, schema='bed').query(f'chrom in {clr.chromnames}')
ctcf['mid'] = (ctcf.end+ctcf.start)//2
ctcf.head()
| chrom | start | end | name | score | strand | mid | |
|---|---|---|---|---|---|---|---|
| 17271 | chr17 | 118485 | 118504 | MA0139.1_CTCF_human | 12.384042 | - | 118494 |
| 17272 | chr17 | 144002 | 144021 | MA0139.1_CTCF_human | 11.542617 | + | 144011 |
| 17273 | chr17 | 163676 | 163695 | MA0139.1_CTCF_human | 5.294219 | - | 163685 |
| 17274 | chr17 | 164711 | 164730 | MA0139.1_CTCF_human | 11.889376 | + | 164720 |
| 17275 | chr17 | 309416 | 309435 | MA0139.1_CTCF_human | 7.879575 | - | 309425 |
import bbi
# Get CTCF ChIP-Seq fold-change over input for genomic regions centered at the positions of the motifs
flank = 250 # Length of flank to one side from the boundary, in basepairs
ctcf_chip_signal = bbi.stackup(
ctcf_fc_file,
ctcf.chrom,
ctcf.mid-flank,
ctcf.mid+flank,
bins=1)
ctcf['FC_score'] = ctcf_chip_signal
ctcf['quartile_score'] = pd.qcut(ctcf['score'], 4, labels=False) + 1
ctcf['quartile_FC_score'] = pd.qcut(ctcf['FC_score'], 4, labels=False) + 1
ctcf['peaks_importance'] = ctcf.apply(
lambda x: 'Top by both scores' if x.quartile_score==4 and x.quartile_FC_score==4 else
'Top by Motif score' if x.quartile_score==4 else
'Top by FC score' if x.quartile_FC_score==4 else 'Ordinary peaks', axis=1
)
# Select the CTCF sites that are in top quartile by both the ChIP-Seq data and motif score
sites = ctcf[ctcf['peaks_importance']=='Top by both scores']\
.sort_values('FC_score', ascending=False)\
.reset_index(drop=True)
sites.tail()
| chrom | start | end | name | score | strand | mid | FC_score | quartile_score | quartile_FC_score | peaks_importance | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 659 | chr17 | 8158938 | 8158957 | MA0139.1_CTCF_human | 13.276979 | - | 8158947 | 25.056849 | 4 | 4 | Top by both scores |
| 660 | chr2 | 176127201 | 176127220 | MA0139.1_CTCF_human | 12.820343 | + | 176127210 | 25.027294 | 4 | 4 | Top by both scores |
| 661 | chr17 | 38322364 | 38322383 | MA0139.1_CTCF_human | 13.534864 | - | 38322373 | 25.010430 | 4 | 4 | Top by both scores |
| 662 | chr2 | 119265336 | 119265355 | MA0139.1_CTCF_human | 13.739862 | - | 119265345 | 24.980141 | 4 | 4 | Top by both scores |
| 663 | chr2 | 118003514 | 118003533 | MA0139.1_CTCF_human | 12.646685 | - | 118003523 | 24.957502 | 4 | 4 | Top by both scores |
sites.to_csv('annotated_ctcf_sites.tsv', sep='\t', index=False, header=False) # Let's save to use in CLI
On-diagonal pileup¶
On-diagonal pileup is the simplest, you need the positions of features (middlepoints of CTCF motifs) and the size of flanks aroung each motif. Coolpuppy will aggregate all snippets around each motif with the specified normalization.
pup = coolpup.pileup(clr, sites, features_format='bed', view_df=hg38_arms, local=True,
flank=300_000, min_diag=0)
INFO:coolpuppy:('chr2_p', 'chr2_p'): 144
INFO:coolpuppy:('chr2_q', 'chr2_q'): 202
INFO:coolpuppy:('chr17_p', 'chr17_p'): 78
INFO:coolpuppy:('chr17_q', 'chr17_q'): 239
INFO:coolpuppy:Total number of piled up windows: 663
This is the general format of output of coolpuppy pileup functions: a pandas dataframe with columns “data” and “n” - “data” contains pileups as numpy arrays, and “n” - number of snippets used to generate this pileup.
Different kinds of pileups calculated in one run are stored as rows, and their groups are annotated in the columns preceding “data”. Since here we didn’t split the data into any groups, there is only one pileup with group “all”
Let’s visualize the average Hi-C map at all strong CTCF sites:
plt.imshow(
np.log10(pup.loc[0, 'data']),
vmax = -1,
vmin = -3.0,
cmap='fall',
interpolation='none')
plt.colorbar(label = 'log10 mean ICed Hi-C')
ticks_pixels = np.linspace(0, flank*2//resolution,5)
ticks_kbp = ((ticks_pixels-ticks_pixels[-1]/2)*resolution//1000).astype(int)
plt.xticks(ticks_pixels, ticks_kbp)
plt.yticks(ticks_pixels, ticks_kbp)
plt.xlabel('relative position, kbp')
plt.ylabel('relative position, kbp')
plt.show()

By-strand pileups¶
Now, we know that orientation of the CTCF site is very important for the interactions it forms. Using coolpuppy, splitting regions by the strand is trivial, expecially using a convenience function:
pup = coolpup.pileup(clr, sites, features_format='bed', view_df=hg38_arms, local=True,
by_strand=True,
flank=300_000, min_diag=0)
INFO:coolpuppy:('chr2_p', 'chr2_p'): 144
INFO:coolpuppy:('chr2_q', 'chr2_q'): 202
INFO:coolpuppy:('chr17_p', 'chr17_p'): 78
INFO:coolpuppy:('chr17_q', 'chr17_q'): 239
INFO:coolpuppy:Total number of piled up windows: 663
pup
| orientation | strand2 | strand1 | data | n | num | clr | resolution | flank | rescale_flank | ... | rescale_size | flip_negative_strand | ignore_diags | store_stripes | nproc | by_window | by_strand | by_distance | groupby | cooler | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | -- | - | - | [[1.8388677780141118, 0.3035714543313729, 0.05... | 326 | [[307, 307, 307, 306, 306, 306, 306, 307, 307,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | 300000 | None | ... | 99 | False | 0 | False | 1 | False | True | False | [] | test |
| 1 | ++ | + | + | [[1.8534148775587997, 0.2993882425820983, 0.05... | 337 | [[325, 324, 324, 322, 322, 320, 320, 321, 320,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | 300000 | None | ... | 99 | False | 0 | False | 1 | False | True | False | [] | test |
| 2 | all | all | all | [[1.846348485849592, 0.30142349774378974, 0.05... | 663 | [[632.0, 631.0, 631.0, 628.0, 628.0, 626.0, 62... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | 300000 | None | ... | 99 | False | 0 | False | 1 | False | True | False | [] | test |
3 rows × 38 columns
Now we can use a convenient seaborn-based function from the plotpup.py subpackage to create a grid of heatmaps based on by-row and/or by-column variable mapping. In this case, we just map two orientations of CTCF sites across columns.
sns.set_theme(font_scale=2, style="ticks")
plotpup.plot(pup,
cols='orientation', col_order=['--', '++'],
score=False, cmap='fall', scale='log', sym=False,
vmin=0.001, vmax=0.1,
height=5)
<seaborn.axisgrid.FacetGrid at 0x7f756cb1b5e0>

Pileups of observed over expected interactions¶
Sometimes you don’t want to include the distance decay P(s) in your pileups. For example, when you make comparison of pileups between experiments and they have different P(s). Even if these differences are slight, they might affect the pileup of raw ICed Hi-C interactions. Moreover, without controlling for it the range of values in the pileup is not very easy to guess before plotting.
In this case, the observed over expected pileup is your choice. To normalize your pileup to the background level of interactions, you can either, prior to running the pileup function, calculate expected interactions for each chromosome arms, or you can generate randomly shifted control regions for each snippet, and divide the final pileup by that control pileup.
Let’s first try the latter. This analysis is particulalry useful for single-cell Hi-C where the data might be too sparse to generate robust per-diagonal expected values.
pup = coolpup.pileup(clr, sites, features_format='bed', view_df=hg38_arms, local=True,
by_strand=True, nshifts=10,
flank=300_000, min_diag=0)
INFO:coolpuppy:('chr2_p', 'chr2_p'): 144
INFO:coolpuppy:('chr2_q', 'chr2_q'): 202
INFO:coolpuppy:('chr17_p', 'chr17_p'): 78
INFO:coolpuppy:('chr17_q', 'chr17_q'): 239
INFO:coolpuppy:Total number of piled up windows: 663
plotpup.plot(pup,
cols='orientation', col_order=['--', '++'],
score=False, cmap='coolwarm', scale='log', sym=True,
vmax=2,
height=5)
<seaborn.axisgrid.FacetGrid at 0x7f756ca16cd0>

As you can see, this strongly highlights the depletion of interactions across the CTCF sites, and enrichment of interactions in a stripe starting from the site.
Now let’s calculate per-diagonal expected level of interactions to repeat the analysis using that.
# Calculate expected interactions for chromosome arms
expected = expected_cis(
clr,
ignore_diags=0,
view_df=hg38_arms,
chunksize=1000000)
expected.to_csv('test_expected_cis.tsv', sep='\t', index=False, header=True) # Let's save to use in CLI
pup = coolpup.pileup(clr, sites, features_format='bed', view_df=hg38_arms, local=True,
expected_df=expected, by_strand=True,
flank=300_000, min_diag=0)
INFO:coolpuppy:('chr2_p', 'chr2_p'): 144
INFO:coolpuppy:('chr2_q', 'chr2_q'): 202
INFO:coolpuppy:('chr17_p', 'chr17_p'): 78
INFO:coolpuppy:('chr17_q', 'chr17_q'): 239
INFO:coolpuppy:Total number of piled up windows: 663
plotpup.plot(pup,
cols='orientation', col_order=['--', '++'],
score=False, cmap='coolwarm', scale='log',
sym=True, vmax=2,
height=5)
<seaborn.axisgrid.FacetGrid at 0x7f756c9d5910>

The result is almost identical!
Instead of splitting two strands into two separate pileups, one can also flip the features on the negative strand. This way a single pileup is created where all features face in the same direction (as if they were on the positive strand). We can also add plot_ticks=True to show the central and flanking coordinates on the bottom of the plot.
pup = coolpup.pileup(clr, sites, features_format='bed', view_df=hg38_arms, local=True,
expected_df=expected, flip_negative_strand=True,
flank=300_000, min_diag=0)
INFO:coolpuppy:('chr2_p', 'chr2_p'): 144
INFO:coolpuppy:('chr2_q', 'chr2_q'): 202
INFO:coolpuppy:('chr17_p', 'chr17_p'): 78
INFO:coolpuppy:('chr17_q', 'chr17_q'): 239
INFO:coolpuppy:Total number of piled up windows: 663
plotpup.plot(pup,
score=False, cmap='coolwarm', scale='log',
sym=True, vmax=2,
height=5, plot_ticks=True)
<seaborn.axisgrid.FacetGrid at 0x7f754d8d11f0>

Arbitrary grouping of snippets for pileups¶
Now, let’s see how our selection of only top CTCF peaks affects the results. We could simply repeat the analysis with the rest of CTCF peaks, but to showcase the power of coolpuppy, we’ll demonstrate how it can be used to generate pileups split be arbitrary categories
pup = coolpup.pileup(clr, ctcf, features_format='bed', view_df=hg38_arms, local=True,
expected_df=expected, flip_negative_strand=True,
groupby=['peaks_importance1'],
flank=300_000, min_diag=0)
# Splitting all snippets into groups based on annotated previously importance of the peaks
# Also flipping negative stranded features as shown above.
INFO:coolpuppy:('chr2_p', 'chr2_p'): 1381
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2221
INFO:coolpuppy:('chr17_p', 'chr17_p'): 548
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1602
INFO:coolpuppy:Total number of piled up windows: 5752
pup
| peaks_importance1 | data | n | num | clr | resolution | flank | rescale_flank | chroms | minshift | ... | rescale_size | flip_negative_strand | ignore_diags | store_stripes | nproc | by_window | by_strand | by_distance | groupby | cooler | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Ordinary peaks | [[1.0699452226771298, 1.0642211188669746, 1.07... | 3536 | [[3432, 3421, 3412, 3413, 3403, 3404, 3402, 33... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | 300000 | None | ['chr2', 'chr17'] | 100000 | ... | 99 | True | 0 | False | 1 | False | False | False | [peaks_importance1] | test |
| 1 | Top by FC score | [[1.0829924317033595, 1.1009490059442415, 1.09... | 778 | [[745, 740, 741, 739, 739, 737, 739, 736, 734,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | 300000 | None | ['chr2', 'chr17'] | 100000 | ... | 99 | True | 0 | False | 1 | False | False | False | [peaks_importance1] | test |
| 2 | Top by Motif score | [[1.0288083600349012, 1.0363397675639456, 1.03... | 775 | [[750, 749, 746, 744, 746, 741, 743, 743, 739,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | 300000 | None | ['chr2', 'chr17'] | 100000 | ... | 99 | True | 0 | False | 1 | False | False | False | [peaks_importance1] | test |
| 3 | Top by both scores | [[1.0773804769093807, 1.0932965447911036, 1.09... | 663 | [[640, 638, 638, 635, 634, 632, 631, 630, 629,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | 300000 | None | ['chr2', 'chr17'] | 100000 | ... | 99 | True | 0 | False | 1 | False | False | False | [peaks_importance1] | test |
| 4 | all | [[1.067003977204076, 1.0686994220484458, 1.072... | 5752 | [[5567.0, 5548.0, 5537.0, 5531.0, 5522.0, 5514... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | 300000 | None | ['chr2', 'chr17'] | 100000 | ... | 99 | True | 0 | False | 1 | False | False | False | [peaks_importance1] | test |
5 rows × 36 columns
fg = plotpup.plot(pup.reset_index(), # Simply resetting the idnex makes the output directly compatible with the plotting function -
# just need to remember there is also a group "all" which you might not want to show
cols='peaks_importance1',
col_order=['Ordinary peaks', 'Top by Motif score',
'Top by FC score', 'Top by both scores'],
score=False, cmap='coolwarm',
scale='log', sym=True, vmax=2,
height=5)

By-distance pileups¶
As it is known, CTCF sites frequently have peaks of Hi-C interactions between them, that indicate chromatin loops. Let’s see at what distances they tend to occur, and let’s see what patterns these regions form at different distance separations and different motif orientations.
Since we generate many more snippets than for local (on-diagonal) pileups, it will take a little longer to run. We can use the nproc argument to use multiprocessing and run it in parallel to speed it up a bit. Note that parallelization requires more RAM, so use with caution.
# Using all strong sites here to make it faster
pup = coolpup.pileup(clr, sites, features_format='bed', view_df=hg38_arms,
expected_df=expected, flip_negative_strand=True,
by_distance=True, by_strand=True, mindist=100_000,
flank=300_000, min_diag=0,
nproc=2
)
# Splitting all snippets into groups based on strand and separation between two sites
INFO:coolpuppy:('chr2_p', 'chr2_p'): 10250
INFO:coolpuppy:('chr17_p', 'chr17_p'): 2959
INFO:coolpuppy:('chr2_q', 'chr2_q'): 20215
INFO:coolpuppy:('chr17_q', 'chr17_q'): 28284
INFO:coolpuppy:Total number of piled up windows: 61708
pup.head()
| separation | orientation | distance_band | strand2 | strand1 | data | n | num | clr | resolution | ... | rescale_size | flip_negative_strand | ignore_diags | store_stripes | nproc | by_window | by_strand | by_distance | groupby | cooler | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0.1Mb-\n0.2Mb | ++ | (100000, 200000) | + | + | [[1.0169219164613472, 0.9600852237815437, 1.01... | 67 | [[64, 66, 66, 65, 66, 65, 63, 65, 65, 65, 65, ... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | ... | 99 | True | 0 | False | 2 | False | True | True | [] | test |
| 1 | 0.2Mb-\n0.4Mb | ++ | (200000, 400000) | + | + | [[0.8621663934782983, 0.8441139544987121, 0.85... | 131 | [[125, 126, 126, 126, 126, 126, 125, 126, 126,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | ... | 99 | True | 0 | False | 2 | False | True | True | [] | test |
| 2 | 0.4Mb-\n0.8Mb | ++ | (400000, 800000) | + | + | [[0.6943248290614478, 0.6951043361705603, 0.68... | 245 | [[236, 236, 236, 236, 236, 236, 236, 236, 236,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | ... | 99 | True | 0 | False | 2 | False | True | True | [] | test |
| 3 | 0.8Mb-\n1.6Mb | ++ | (800000, 1600000) | + | + | [[0.6474314279066417, 0.6349530137034375, 0.68... | 425 | [[390, 390, 390, 390, 394, 389, 389, 393, 393,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | ... | 99 | True | 0 | False | 2 | False | True | True | [] | test |
| 4 | 1.6Mb-\n3.2Mb | ++ | (1600000, 3200000) | + | + | [[0.8110724673135341, 0.8450568161843092, 0.79... | 789 | [[727, 727, 727, 727, 732, 727, 718, 736, 734,... | /gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy... | 10000 | ... | 99 | True | 0 | False | 2 | False | True | True | [] | test |
5 rows × 40 columns
fg = plotpup.plot(pup, rows='orientation', cols='separation',
row_order=['-+', '--', '++', '+-'],
score=False, cmap='coolwarm', scale='log', sym=True, vmax=3,
height=3)

Note that since CTCF sites are preferentially found in the A compartment, the expected level of interactions at different distances varies, generating different background interaction levels. We can artificially fix that by normalizing each pileup by the average interactions in the top-left and bottom-right corners.
fg = plotpup.plot(pup, rows='orientation', cols='separation',
row_order=['-+', '--', '++', '+-'],
score=False, cmap='coolwarm', scale='log', sym=True, vmax=3,
norm_corners=10,
height=3)

If you want to actually modify the data in your dataframe to normalize to the corners and not just apply it for vizualisation, you can do that explicitly:
pup['data'] = pup['data'].apply(numutils.norm_cis, i=10)
A good idea to give some more quantitative information about the level of enrichment of interactions in the center of the pileup is to just label the average value of the few central pixels of the heatmap. The simplest way is to use the argument score:
fg = plotpup.plot(pup, rows='orientation', cols='separation',
row_order=['-+', '--', '++', '+-'],
score=True,
cmap='coolwarm', scale='log', sym=True, vmax=3,
height=3)

Dividing pileups¶
Sometimes you may want to compare two pileups directly and plot the result of the division between them. For this we can use the divide_pups function. Let’s look at all CTCF interactions between 100 kb and 1 Mb by motif orientation.
pup = coolpup.pileup(clr, sites, features_format='bed', view_df=hg38_arms,
expected_df=expected,
by_strand=True, mindist=100_000, maxdist=1_000_000,
flank=300_000, nproc=2)
INFO:coolpuppy:('chr2_p', 'chr2_p'): 287
INFO:coolpuppy:('chr2_q', 'chr2_q'): 522
INFO:coolpuppy:('chr17_p', 'chr17_p'): 262
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1235
INFO:coolpuppy:Total number of piled up windows: 2306
plotpup.plot(pup,
cols="orientation",
col_order=["-+", "--", "++", "+-"],
score=False, cmap='coolwarm', scale='log',
sym=True, vmax=2,
height=2)
<seaborn.axisgrid.FacetGrid at 0x7f7631b5a850>

Let’s compare the ++ to the – CTCF motif orientation pileups. First, we have to create two separate pileup dataframes from the strand-separated pileups. Alternatively, you could generate two new pileups and store them. Importantly, the two pileups cannot differ with regards to the columns they contain and the resolution, flank size etc. they’ve been generated using.
pup_plus = pup.loc[pup["orientation"]=="++"].drop(columns=["strand1", "strand2", "orientation"])
pup_minus = pup.loc[pup["orientation"]=="--"].drop(columns=["strand1", "strand2", "orientation"])
pup_divide = divide_pups(pup_plus, pup_minus)
plotpup.plot(pup_divide,
score=False, cmap='PuOr_r', scale='log',
sym=True, height=4)
<seaborn.axisgrid.FacetGrid at 0x7f7631cebe50>

Stripe stackups¶
Oftentimes, as seen in the examples above, the interactions between regions are not just focal, but seen as stripes with enrichment along the vertical/horizontal axis from one or both of the anchor points. In the CTCF pileups from above we see a very strong corner stripe between +- sites, so let’s try to plot these individual stripes. Below is a schematic of what is meant by the different types of stripes.

We first have store this information when generating the pileup using the store_stripes=True argument which will add the columns vertical_stripe, and horizontal_stripe to the output. These are used to calculate the corner stripe in the plotting function.
pup = coolpup.pileup(clr, sites, features_format='bed', view_df=hg38_arms,
expected_df=expected,
by_strand=True, mindist=100_000, maxdist=1_000_000,
flank=300_000, nproc=2,
store_stripes=True)
INFO:coolpuppy:('chr2_p', 'chr2_p'): 287
INFO:coolpuppy:('chr2_q', 'chr2_q'): 522
INFO:coolpuppy:('chr17_p', 'chr17_p'): 262
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1235
INFO:coolpuppy:Total number of piled up windows: 2306
We can plot the stripes using the the plot_stripes function
sns.set(font_scale = 1.3, style="ticks")
plotpup.plot_stripes(pup.loc[(pup["strand1"] == "+") &
(pup["strand2"] == "-"),:],
vmax=10, height=4,
stripe="vertical_stripe", stripe_sort="sum",
plot_ticks=True)
<seaborn.axisgrid.FacetGrid at 0x7f76319c6b20>

Each line of the above plot represents the “corner stripe” between two regions. These pairs are sorted by the sum of the stripe by default, but we can also sort them by the central pixel, i.e. the pixel where the two regions of interest interact, with the stripe_sort argument. We can further save the pairs in the sorted order using out_sorted_bedpe. This file can then be used to inspect individual pairs with high contact frequencies. We can also add a lineplot with the average signal above the stripes using lineplot (note that this only works for single stripe plots.)
plotpup.plot_stripes(pup.loc[(pup["strand1"] == "+") &
(pup["strand2"] == "-"),:],
vmax=10, height=4,
stripe="corner_stripe", plot_ticks=True,
stripe_sort="center_pixel",
out_sorted_bedpe="CTCF_+-_sorted_centerpixel.bedpe",
lineplot=True)
<seaborn.axisgrid.FacetGrid at 0x7f754dc03cd0>

Rescaling¶
Pileups can also be rescaled to visualise enrichment within regions of interests of different sizes using rescale=True. The rescale_flank value represents how large the flanks are compared to the region of interest, where 1 is equal in size and for example 3 will be three times the size. The number of pixels in the final plot after rescaling is set with rescale_size. Let’s try this for B compartment interactions.
# Load cooler at 1 Mb resolution
clr_1Mb = cooler.Cooler(f'{cool_file}::/resolutions/1000000')
# Calculate eigenvectors
cis_eigs = cooltools.eigs_cis(clr_1Mb, n_eigs=3)
eigenvector_track = cis_eigs[1][['chrom','start','end','E1']]
# Extract B compartments
B_compartments = bioframe.merge(eigenvector_track[eigenvector_track["E1"] < 0], min_dist=0)
# Let's save to use it in CLI
B_compartments[["chrom", "start", "end"]].to_csv("B_compartments.bed", sep="\t", header=None, index=False)
pup = coolpup.pileup(clr, B_compartments, features_format='bed', view_df=hg38_arms,
expected_df=expected,
rescale=True, rescale_flank=1, rescale_size=99,
flank=300_000, nproc=2)
INFO:coolpuppy:Rescaling with rescale_flank = 1 to 99x99 pixels
INFO:coolpuppy:('chr2_p', 'chr2_p'): 36
INFO:coolpuppy:('chr17_p', 'chr17_p'): 6
INFO:coolpuppy:('chr17_q', 'chr17_q'): 21
INFO:coolpuppy:('chr2_q', 'chr2_q'): 153
INFO:coolpuppy:Total number of piled up windows: 216
fg = plotpup.plot(pup,
score=False, cmap='coolwarm', scale='log',
sym=True, vmax=2, height=3)

Trans (inter-chromosomal) pileups¶
We can also perform pileups between regions on different chromosomes. We will try this for insulation score boundaries (TAD boundaries), first for cis (within chromosomes) and then for trans (between chromosomes).
# Call insulation score at windows of 50 kb
insulation_table = cooltools.insulation(clr, [50000], verbose=True)
# Select strong boundaries
strong_boundaries = insulation_table.loc[insulation_table["boundary_strength_50000"] > 1.5, :]
# Let's save to use it in CLI
strong_boundaries[["chrom", "start", "end"]].to_csv("strong_boundaries.bed", sep="\t", header=None, index=False)
INFO:root:Processing region chr2
INFO:root:Processing region chr17
pup = coolpup.pileup(clr, strong_boundaries, features_format='bed', view_df=hg38_arms,
expected_df=expected,
flank=300_000, nproc=2)
INFO:coolpuppy:('chr2_p', 'chr2_p'): 807
INFO:coolpuppy:('chr17_p', 'chr17_p'): 99
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2262
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1354
INFO:coolpuppy:Total number of piled up windows: 4522
plotpup.plot(pup,
score=False, cmap='coolwarm', scale='log',
sym=True, vmax=2,
height=3)
<seaborn.axisgrid.FacetGrid at 0x7f763151f8e0>

Here we can see the boundary pileups within chromosome arms where interactions are depleted at the boundaries. To perform the same analysis between chromosomes, we first need to generate a new expected file (or use shifted controls) and then run the analysis with trans=True.
# Calculate expected interactions between chromosomes
trans_expected = expected_trans(
clr,
chunksize=1000000)
trans_expected
| region1 | region2 | n_valid | count.sum | balanced.sum | count.avg | balanced.avg | |
|---|---|---|---|---|---|---|---|
| 0 | chr2 | chr17 | 169848938 | 1303548.0 | 206.059958 | 0.007675 | 0.000001 |
pup = coolpup.pileup(clr, strong_boundaries, features_format='bed',
expected_df=trans_expected,
trans=True,
flank=300_000, nproc=2)
/gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy_trans/coolpuppy/coolpuppy/coolpup.py:2107: UserWarning: Ignoring maxdist when using trans
CC = CoordCreator(
INFO:coolpuppy:('chr2', 'chr17'): 7412
INFO:coolpuppy:Total number of piled up windows: 7412
plotpup.plot(pup,
score=False, cmap='coolwarm', scale='log',
sym=True, vmax=2,
height=3)
<seaborn.axisgrid.FacetGrid at 0x7f7630d5ba90>

Here we can see that boundaries are depleted in interactions also between chromosomes.
Coolpuppy CLI walkthrough notebook¶
Please first see the python API examples for a more detailed introduction. Here we will reproduce all of the plots from the API notebook, but only using CLI! Note that, however, the API tutorial saves some files used in the commands here which would be tricky to obtain using CLI tools only.
# We can use this function to display a file within the notebook
from IPython.display import Image
import cooltools
# Downloading test data for pileups
# cache = True will doanload the data only if it was not previously downloaded
# data_dir="./" will force download to the current directory
cool_file = cooltools.download_data("HFF_MicroC", cache=True, data_dir='./')
ctcf_peaks_file = cooltools.download_data("HFF_CTCF_binding", cache=True, data_dir='./')
ctcf_fc_file = cooltools.download_data("HFF_CTCF_fc", cache=True, data_dir='./')
Simple local pileup¶
First a simple local pileup around all CTCF sites. This command will save the pileup in a hdf5-based file together with all parameters that were used when running it.
!coolpup.py test.mcool::resolutions/10000 test_CTCF.bed.gz \
--features_format bed --local --nshifts 0 \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 \
--outname local_CTCF_pileup_nonorm.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 1381
INFO:coolpuppy:('chr17_p', 'chr17_p'): 548
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2221
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1602
INFO:coolpuppy:Total number of piled up windows: 5752
INFO:coolpuppy:Saved output to local_CTCF_pileup_nonorm.clpy
This is the plotting command, which in this case simply takes the path to the file we just produced, the output path, and some arguments to control the esthetics of thefigure.
!plotpup.py --cmap fall --vmax 0.1 --vmin 0.001 \
--no_score \
--input_pups local_CTCF_pileup_nonorm.clpy \
--output local_CTCF_pileup_nonorm.png
Image('local_CTCF_pileup_nonorm.png')
INFO:coolpuppy:Can't set both vmin and vmax and get symmetrical scale. Plotting non-symmetrical
INFO:coolpuppy:Saved output to local_CTCF_pileup_nonorm.png

Pileups by strand¶
Now let’s split the pileup in two, based on the strands of CTCF sites. There is a simple “preset” for that, you simply need to add --by_strand argument.
!coolpup.py test.mcool::resolutions/10000 test_CTCF.bed.gz \
--features_format bed --local --nshifts 0 \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 \
--by-strand \
--outname local_CTCF_pileup_bystrand_nonorm.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 1381
INFO:coolpuppy:('chr17_p', 'chr17_p'): 548
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2221
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1602
INFO:coolpuppy:Total number of piled up windows: 5752
INFO:coolpuppy:Saved output to local_CTCF_pileup_bystrand_nonorm.clpy
!plotpup.py --cols orientation \
--col_order "-- ++" \
--cmap fall --vmax 0.1 --vmin 0.001 \
--no_score \
--input_pups local_CTCF_pileup_bystrand_nonorm.clpy \
--output local_CTCF_pileup_bystrand_nonorm.png
Image('local_CTCF_pileup_bystrand_nonorm.png')
INFO:coolpuppy:Can't set both vmin and vmax and get symmetrical scale. Plotting non-symmetrical
INFO:coolpuppy:Saved output to local_CTCF_pileup_bystrand_nonorm.png

Normalization to background interaction level¶
Random shifts¶
Now let’s repeat the above, but also normalize the pileups to the decay of contact probability with separation. You can either use the randomly shifted control regions (here) or a global expected level of interactions calculated for the whole chromosome arm (see below).
!coolpup.py test.mcool::resolutions/10000 test_CTCF.bed.gz \
--features_format bed --local --by_strand --nshifts 1 \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 \
--outname local_CTCF_pileup_bystrand_1shift.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 1381
INFO:coolpuppy:('chr17_p', 'chr17_p'): 548
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2221
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1602
INFO:coolpuppy:Total number of piled up windows: 5752
INFO:coolpuppy:Saved output to local_CTCF_pileup_bystrand_1shift.clpy
!plotpup.py --cols orientation \
--col_order "-- ++" \
--no_score \
--input_pups local_CTCF_pileup_bystrand_1shift.clpy \
--output local_CTCF_pileup_bystrand_1shift.png
Image('local_CTCF_pileup_bystrand_1shift.png')
INFO:coolpuppy:Saved output to local_CTCF_pileup_bystrand_1shift.png

Chromosome arm-wide expected normalization¶
While we computed the expected using cooltools python API in the API notebook, here is the CLI version of the same process, with 2 cores.
!cooltools expected-cis --view hg38_arms.bed -p 2 -o test_expected_cis.tsv test.mcool::resolutions/10000
This is a little faster than using random shifts, and in most cases results are very similar. Therefore when a cooler file is used multiple times, it’s beneficial to use this approach.
!coolpup.py test.mcool::resolutions/10000 test_CTCF.bed.gz \
--features_format bed --local --by_strand --expected test_expected_cis.tsv \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 \
--outname local_CTCF_pileup_bystrand_expected.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 1381
INFO:coolpuppy:('chr17_p', 'chr17_p'): 548
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2221
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1602
INFO:coolpuppy:Total number of piled up windows: 5752
INFO:coolpuppy:Saved output to local_CTCF_pileup_bystrand_expected.clpy
!plotpup.py --cols orientation \
--col_order "-- ++" \
--no_score \
--input_pups local_CTCF_pileup_bystrand_expected.clpy \
--output local_CTCF_pileup_bystrand_expected.png
Image('local_CTCF_pileup_bystrand_expected.png')
INFO:coolpuppy:Saved output to local_CTCF_pileup_bystrand_expected.png

Instead of splitting two strands into two separate pileups, one can also flip the features on the negative strand using --flip_negative_strand. This way a single pileup is created where all features face in the same direction (as if they were on the positive strand). We can also add --plot_ticks to show the central and flanking coordinates on the bottom of the plot.
!coolpup.py test.mcool::resolutions/10000 test_CTCF.bed.gz \
--features_format bed --local --expected test_expected_cis.tsv \
--flip_negative_strand --ignore_diags 0 --view hg38_arms.bed --flank 300000 \
--outname local_CTCF_pileup_flipped_expected.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 1381
INFO:coolpuppy:('chr17_p', 'chr17_p'): 548
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2221
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1602
INFO:coolpuppy:Total number of piled up windows: 5752
INFO:coolpuppy:Saved output to local_CTCF_pileup_flipped_expected.clpy
!plotpup.py --plot_ticks --height 1.5 \
--no_score \
--input_pups local_CTCF_pileup_flipped_expected.clpy \
--output local_CTCF_pileup_flipped_expected.png
Image('local_CTCF_pileup_flipped_expected.png')
INFO:coolpuppy:Saved output to local_CTCF_pileup_flipped_expected.png

Arbitrary grouping of snippets for pileups¶
Now, let’s see if selecting different strength CTCF peaks affects the results. To showcase the power of coolpuppy, we’ll demonstrate how it can be used to generate pileups split be arbitrary categories using groupby. Note that the input peak file needs to include column names in order to use groupby
import pandas as pd
import bioframe
ctcf = bioframe.read_table(ctcf_peaks_file, schema='bed')
ctcf['quartile_score'] = pd.qcut(ctcf['score'], 4, labels=False) + 1
ctcf.to_csv('ctcf_sites.tsv', sep='\t', index=False, header=True)
!coolpup.py test.mcool::resolutions/10000 ctcf_sites.tsv \
--features_format bed --local --expected test_expected_cis.tsv \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 \
--flip_negative_strand --groupby quartile_score1 \
--outname groupby_score_CTCF_pileup_expected.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 1381
INFO:coolpuppy:('chr17_p', 'chr17_p'): 548
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2221
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1602
INFO:coolpuppy:Total number of piled up windows: 5752
INFO:coolpuppy:Saved output to groupby_score_CTCF_pileup_expected.clpy
!plotpup.py --plot_ticks --height 1.5 \
--no_score --cols quartile_score1 \
--col_order '1 2 3 4' \
--input_pups groupby_score_CTCF_pileup_expected.clpy \
--output groupby_score_CTCF_pileup_expected.png
Image('groupby_score_CTCF_pileup_expected.png')
INFO:coolpuppy:Saved output to groupby_score_CTCF_pileup_expected.png

By-distance pileups¶
Now we can add another layer of complexity: look at distal interactions betwwen CTCF sites, and split all snippets by their distance.
We use the file that we saved in the python API notebook that contains the annotation of site strength. coolpup.py can accept the coordinate input from stdin, so we can filter that file on the fly using awk, and this way we can use only the strong CTCF sites.
# This command will take a bit longer to run, since it's averaging over a much larger number of snippets
!cat annotated_ctcf_sites.tsv | awk -F'\t' '($11 == "Top by both scores")' | coolpup.py test.mcool::resolutions/10000 - \
--features_format bed --by_distance --by_strand --expected test_expected_cis.tsv \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 --mindist 100000 --maxdist 102400000 \
--outname bydistance_CTCF_pileup_bystrand_expected.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 10250
INFO:coolpuppy:('chr17_p', 'chr17_p'): 2959
INFO:coolpuppy:('chr2_q', 'chr2_q'): 15938
INFO:coolpuppy:('chr17_q', 'chr17_q'): 28284
INFO:coolpuppy:Total number of piled up windows: 57431
INFO:coolpuppy:Saved output to bydistance_CTCF_pileup_bystrand_expected.clpy
# "separation" is created when the pileups are created by distance, and plotpup.py
# always plots them in the order of increasing distance
# We need to specify the order of rows, otherise it's not guaranteed
!plotpup.py --cols separation \
--rows orientation \
--row_order "-+ -- ++ +-" \
--vmax 3 \
--input_pups bydistance_CTCF_pileup_bystrand_expected.clpy \
--output bydistance_CTCF_pileup_bystrand_expected.png
Image('bydistance_CTCF_pileup_bystrand_expected.png')
INFO:coolpuppy:Saved output to bydistance_CTCF_pileup_bystrand_expected.png

Now we can also normalize each pileup to the average value in its top-left and bottom-right corners to remove the variation in background level of interactions
!plotpup.py --cols separation \
--rows orientation \
--row_order "-+ -- ++ +-"\
--vmax 3 --norm_corners 10 \
--input_pups bydistance_CTCF_pileup_bystrand_expected.clpy \
--output bydistance_CTCF_pileup_bystrand_expected_corner_norm.png
Image('bydistance_CTCF_pileup_bystrand_expected_corner_norm.png')
INFO:coolpuppy:Saved output to bydistance_CTCF_pileup_bystrand_expected_corner_norm.png

Dividing pileups¶
Sometimes you may want to compare two pileups directly and plot the result of the division between them. For this we can use the dividepups.py command. Let’s look at all CTCF interactions between 100 kb and 1 Mb by motif orientation.
!cat annotated_ctcf_sites.tsv | awk -F'\t' '($11 == "Top by both scores")' | coolpup.py test.mcool::resolutions/10000 - \
--features_format bed --by_strand --expected test_expected_cis.tsv \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 --mindist 100000 --maxdist 1000000 \
--outname CTCF_pileup_bystrand_expected.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 287
INFO:coolpuppy:('chr2_q', 'chr2_q'): 522
INFO:coolpuppy:('chr17_p', 'chr17_p'): 262
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1235
INFO:coolpuppy:Total number of piled up windows: 2306
INFO:coolpuppy:Saved output to CTCF_pileup_bystrand_expected.clpy
!plotpup.py --cols orientation \
--col_order "-+ -- ++ +-" \
--vmax 2 \
--no_score \
--input_pups CTCF_pileup_bystrand_expected.clpy \
--output CTCF_pileup_bystrand_expected.png
Image('CTCF_pileup_bystrand_expected.png')
INFO:coolpuppy:Saved output to CTCF_pileup_bystrand_expected.png

Let’s compare the ++ to the – CTCF motif orientation pileups. First, we have to generate two new pileups for each of the orientations. Importantly, the two pileups cannot differ with regards to the columns they contain and the resolution, flank size etc. they’ve been generated using.
!cat annotated_ctcf_sites.tsv | awk -F'\t' '($11 == "Top by both scores") && ($6 == "+")' | coolpup.py test.mcool::resolutions/10000 - \
--features_format bed --expected test_expected_cis.tsv \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 --mindist 100000 --maxdist 1000000 \
--outname CTCF_pileup_plusstrand_expected.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 78
INFO:coolpuppy:('chr2_q', 'chr2_q'): 122
INFO:coolpuppy:('chr17_p', 'chr17_p'): 67
INFO:coolpuppy:('chr17_q', 'chr17_q'): 293
INFO:coolpuppy:Total number of piled up windows: 560
INFO:coolpuppy:Saved output to CTCF_pileup_plusstrand_expected.clpy
!cat annotated_ctcf_sites.tsv | awk -F'\t' '($11 == "Top by both scores") && ($6 == "-")' | coolpup.py test.mcool::resolutions/10000 - \
--features_format bed --expected test_expected_cis.tsv \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 --mindist 100000 --maxdist 1000000 \
--outname CTCF_pileup_minusstrand_expected.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 62
INFO:coolpuppy:('chr2_q', 'chr2_q'): 118
INFO:coolpuppy:('chr17_p', 'chr17_p'): 53
INFO:coolpuppy:('chr17_q', 'chr17_q'): 324
INFO:coolpuppy:Total number of piled up windows: 557
INFO:coolpuppy:Saved output to CTCF_pileup_minusstrand_expected.clpy
Now we will generate a new pileup of the ratio between the two
!dividepups.py CTCF_pileup_plusstrand_expected.clpy CTCF_pileup_minusstrand_expected.clpy \
--outname CTCF_pileup_plus_over_minus.clpy
INFO:root:Namespace(input_pups=['CTCF_pileup_plusstrand_expected.clpy', 'CTCF_pileup_minusstrand_expected.clpy'], outname='CTCF_pileup_plus_over_minus.clpy')
INFO:root:Saved output to CTCF_pileup_plus_over_minus.clpy
!plotpup.py --no_score --cmap PuOr_r \
--input_pups CTCF_pileup_plus_over_minus.clpy \
--output CTCF_pileup_plus_over_minus.png
Image('CTCF_pileup_plus_over_minus.png')
INFO:coolpuppy:Saved output to CTCF_pileup_plus_over_minus.png

If you want to quickly plot the division of two pileups without saving the intermediate, this can also be done directly in plotpup.py with the argument --divide_pups
Stripe stackups¶
Oftentimes, as seen in the examples above, the interactions between regions are not just focal, but seen as stripes with enrichment along the vertical/horizontal axis from one or both of the anchor points. In the CTCF pileups from above we see a very strong corner stripe between +- sites, so let’s try to plot these individual stripes. Below is a schematic of what is meant by the different types of stripes.
!cat annotated_ctcf_sites.tsv | awk -F'\t' '($11 == "Top by both scores")' | coolpup.py test.mcool::resolutions/10000 - \
--features_format bed --by_strand --expected test_expected_cis.tsv \
--ignore_diags 0 --view hg38_arms.bed --flank 300000 --mindist 100000 --maxdist 1000000 \
--outname bystrand_CTCF_pileup_bystrand_expected_stripes.clpy --nproc 2 --store_stripes
INFO:coolpuppy:('chr2_p', 'chr2_p'): 287
INFO:coolpuppy:('chr2_q', 'chr2_q'): 522
INFO:coolpuppy:('chr17_p', 'chr17_p'): 262
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1235
INFO:coolpuppy:Total number of piled up windows: 2306
INFO:coolpuppy:Saved output to bystrand_CTCF_pileup_bystrand_expected_stripes.clpy
In the pileups we see a very strong corner stripe between +- sites, so let’s try to plot these individual stripes using the --stripe argument. Below is a schematic of what is meant by the different types of stripes.
!plotpup.py --rows orientation \
--row_order "+-" --stripe corner_stripe \
--vmax 10 --height 1.5 --font_scale 0.75 --plot_ticks \
--input_pups bystrand_CTCF_pileup_bystrand_expected_stripes.clpy \
--output bystrand_CTCF_pileup_+-_expected_cornerstripe.png
Image('bystrand_CTCF_pileup_+-_expected_cornerstripe.png')
INFO:coolpuppy:Saved output to bystrand_CTCF_pileup_+-_expected_cornerstripe.png

Each line of the above plot represents the “corner stripe” between two regions. These pairs are sorted by the sum of the stripe by default, but we can also sort them by the central pixel, i.e. the pixel where the two regions of interest interact, with the --stripe_sort argument. We can further save the pairs in the sorted order using --out_sorted_bedpe. This file can then be used to inspect individual pairs with high contact frequencies. We can also add a lineplot with the average signal above the stripes using --lineplot (note that this only works for single stripe plots.)
!plotpup.py --rows orientation \
--row_order "+-" --stripe corner_stripe \
--vmax 10 --height 1.5 --font_scale 0.75 --plot_ticks \
--input_pups bystrand_CTCF_pileup_bystrand_expected_stripes.clpy \
--output bystrand_CTCF_pileup_+-_expected_cornerstripe_centersort.png \
--stripe_sort center_pixel --out_sorted_bedpe CTCF_+-_sorted_centerpixel.bedpe \
--lineplot
Image('bystrand_CTCF_pileup_+-_expected_cornerstripe_centersort.png')
INFO:coolpuppy:Saved output to bystrand_CTCF_pileup_+-_expected_cornerstripe_centersort.png

Rescaling¶
Pileups can also be rescaled to visualise enrichment within regions of interests of different sizes using --rescale. The --rescale_flank value represents how large the flanks are compared to the region of interest, where 1 is equal in size and for example 3 will be a three times the size. The number of pixels in the final plot after rescaling is set with --rescale_size. Let’s try this for B compartment interactions.
!coolpup.py test.mcool::resolutions/10000 B_compartments.bed \
--features_format bed --expected test_expected_cis.tsv \
--ignore_diags 0 --view hg38_arms.bed \
--rescale --rescale_flank 1 --rescale_size 99 \
--outname B_compartment_pileup_rescaled_expected.clpy --nproc 2
INFO:coolpuppy:Rescaling with rescale_flank = 1.0 to 99x99 pixels
INFO:coolpuppy:('chr2_p', 'chr2_p'): 36
INFO:coolpuppy:('chr17_p', 'chr17_p'): 6
INFO:coolpuppy:('chr17_q', 'chr17_q'): 21
INFO:coolpuppy:('chr2_q', 'chr2_q'): 153
INFO:coolpuppy:Total number of piled up windows: 216
INFO:coolpuppy:Saved output to B_compartment_pileup_rescaled_expected.clpy
!plotpup.py --vmax 2 --no_score \
--input_pups B_compartment_pileup_rescaled_expected.clpy \
--output B_compartment_pileup_rescaled_expected.png
Image('B_compartment_pileup_rescaled_expected.png')
INFO:coolpuppy:Saved output to B_compartment_pileup_rescaled_expected.png

Trans (inter-chromosomal) pileups¶
We can also perform pileups between regions on different chromosomes. We will try this for insulation score boundaries (TAD boundaries), first for cis (within chromosomes) and then for trans (between chromosomes).
!coolpup.py test.mcool::resolutions/10000 strong_boundaries.bed \
--features_format bed --expected test_expected_cis.tsv \
--view hg38_arms.bed --flank 300000 \
--outname strong_boundaries_pileup_cis_expected.clpy --nproc 2
INFO:coolpuppy:('chr2_p', 'chr2_p'): 807
INFO:coolpuppy:('chr17_p', 'chr17_p'): 99
INFO:coolpuppy:('chr2_q', 'chr2_q'): 2262
INFO:coolpuppy:('chr17_q', 'chr17_q'): 1354
INFO:coolpuppy:Total number of piled up windows: 4522
INFO:coolpuppy:Saved output to strong_boundaries_pileup_cis_expected.clpy
!plotpup.py --vmax 2 --no_score \
--input_pups strong_boundaries_pileup_cis_expected.clpy \
--output strong_boundaries_pileup_cis_expected.png
Image('strong_boundaries_pileup_cis_expected.png')
INFO:coolpuppy:Saved output to strong_boundaries_pileup_cis_expected.png

Here we can see the boundary pileups within chromosome arms where interactions are depleted at the boundaries. To perform the same analysis between chromosomes, we need to use an expected file generated for trans (or use shifted controls) and then run the analysis with --trans.
!cooltools expected-trans -p 2 -o test_expected_trans.tsv test.mcool::resolutions/10000
!coolpup.py test.mcool::resolutions/10000 strong_boundaries.bed \
--features_format bed --expected test_expected_trans.tsv \
--flank 300000 --trans \
--outname strong_boundaries_pileup_trans_expected.clpy --nproc 2
/gpfs/igmmfs01/eddie/wendy-lab/elias/coolpuppy_trans/coolpuppy/coolpuppy/coolpup.py:2115: UserWarning: Ignoring maxdist when using trans
CC = CoordCreator(
INFO:coolpuppy:('chr2', 'chr17'): 7412
INFO:coolpuppy:Total number of piled up windows: 7412
INFO:coolpuppy:Saved output to strong_boundaries_pileup_trans_expected.clpy
!plotpup.py --vmax 2 --no_score \
--input_pups strong_boundaries_pileup_trans_expected.clpy \
--output strong_boundaries_pileup_trans_expected.png
Image('strong_boundaries_pileup_trans_expected.png')
INFO:coolpuppy:Saved output to strong_boundaries_pileup_trans_expected.png

Here we can see that boundaries are depleted in interactions also between chromosomes.
Distribution of TAD strength scores¶
Using some advanced techniques, it’s possible to calculate an arbitrary score for each snippet that contributed to the final pileup, and save those values within the pileup dataframe. This can be used to investigate whether the contribution features are all similar, or only some outliers cause enrichment.
Here as an example, we can calculate and store the TAD strength for each TAD that was averaged.
# If you are a developer, you may want to reload the packages on a fly.
# Jupyter has a magic for this particular purpose:
%load_ext autoreload
%autoreload 2
# import standard python libraries
import matplotlib as mpl
%matplotlib inline
mpl.rcParams['figure.dpi'] = 96
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
import seaborn as sns
# import libraries for biological data analysis
from coolpuppy import coolpup
from coolpuppy.lib.numutils import get_domain_score
from coolpuppy.lib.puputils import accumulate_values
from coolpuppy import plotpup
import cooler
import bioframe
import cooltools
from cooltools.lib import io
from cooltools import insulation, expected_cis
from cooltools.lib import plotting
# Downloading test data for pileups
# cache = True will doanload the data only if it was not previously downloaded
# data_dir="./" will force download to the current directory
cool_file = cooltools.download_data("HFF_MicroC", cache=True, data_dir='./')
# Open cool file with Micro-C data:
clr = cooler.Cooler(f'{cool_file}::/resolutions/10000')
# Set up selected data resolution:
resolution = 10000
# Use bioframe to fetch the genomic features from the UCSC.
hg38_chromsizes = bioframe.fetch_chromsizes('hg38')
hg38_cens = bioframe.fetch_centromeres('hg38')
hg38_arms = bioframe.make_chromarms(hg38_chromsizes, hg38_cens)
# Select only chromosomes that are present in the cooler.
# This step is typically not required! we call it only because the test data are reduced.
hg38_arms = hg38_arms.set_index("chrom").loc[clr.chromnames].reset_index()
# call this to automaticly assign names to chromosomal arms:
hg38_arms = bioframe.make_viewframe(hg38_arms)
# Calculate expected interactions for chromosome arms
expected = expected_cis(
clr,
ignore_diags=0,
view_df=hg38_arms,
chunksize=1000000)
First we need to generate coordinates of TADs. It’s quite simple using cooltools.insulation: we get coordinates of strongly insulating regions, which likely correspond to TAD boundaries. Then we just need to combine consecutive boundaries, filter out super long domains, and we have a list of TAD coordiantes.
insul_df = insulation(clr, window_bp=[100000], view_df=hg38_arms, nproc=4,)
insul_df
| chrom | start | end | region | is_bad_bin | log2_insulation_score_100000 | n_valid_pixels_100000 | boundary_strength_100000 | is_boundary_100000 | |
|---|---|---|---|---|---|---|---|---|---|
| 0 | chr2 | 0 | 10000 | chr2_p | True | NaN | 0.0 | NaN | False |
| 1 | chr2 | 10000 | 20000 | chr2_p | False | 0.692051 | 8.0 | NaN | False |
| 2 | chr2 | 20000 | 30000 | chr2_p | False | 0.760561 | 17.0 | NaN | False |
| 3 | chr2 | 30000 | 40000 | chr2_p | False | 0.766698 | 27.0 | NaN | False |
| 4 | chr2 | 40000 | 50000 | chr2_p | False | 0.674906 | 37.0 | NaN | False |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| 32541 | chr17 | 83210000 | 83220000 | chr17_q | True | NaN | 0.0 | NaN | False |
| 32542 | chr17 | 83220000 | 83230000 | chr17_q | True | NaN | 0.0 | NaN | False |
| 32543 | chr17 | 83230000 | 83240000 | chr17_q | True | NaN | 0.0 | NaN | False |
| 32544 | chr17 | 83240000 | 83250000 | chr17_q | True | NaN | 0.0 | NaN | False |
| 32545 | chr17 | 83250000 | 83257441 | chr17_q | True | NaN | 0.0 | NaN | False |
32552 rows × 9 columns
# A useful function to combine insulation score valleys into TADs and filter out very long "TADs"
def make_tads(insul_df, maxlen=1_500_000):
tads = (
insul_df.groupby("chrom")
.apply(
lambda x: pd.concat(
[x[:-1].reset_index(drop=True), x[1:].reset_index(drop=True)],
axis=1,
ignore_index=True,
)
)
.reset_index(drop=True)
)
tads.columns = [["chrom1", "start1", "end1", "chrom2", "start2", "end2"]]
tads.columns = tads.columns.get_level_values(0)
tads = tads[
(tads["start2"] - tads["start1"]) <= maxlen
].reset_index(drop=True)
tads["start"] = (tads["start1"] + tads["end1"]) // 2
tads["end"] = (tads["start2"] + tads["end2"]) // 2
tads = tads[["chrom1", "start", "end"]]
tads.columns = ['chrom', 'start', 'end']
return tads
Getting TAD coordinates:
tads = make_tads(insul_df[insul_df['is_boundary_100000']][['chrom', 'start', 'end']])
Define a helper function to store domain scores within each snippet:
def add_domain_score(snippet):
snippet['domain_score'] = get_domain_score(snippet['data']) # Calculates domain score for each snippet according to Flyamer et al., 2017
return snippet
Another helper function to save domain scores when combining snippets into a pileup:
def extra_sum_func(dict1, dict2):
return accumulate_values(dict1, dict2, 'domain_score')
Here we use the low-level coolpuppy API, including the helper functions we defined above:
cc = coolpup.CoordCreator(tads, resolution=10000, features_format='bed', local=True, rescale_flank=1)
pu = coolpup.PileUpper(clr, cc, expected=expected, view_df=hg38_arms, ignore_diags=0, rescale_size=99, rescale=True)
pup = pu.pileupsWithControl(postprocess_func=add_domain_score, # Any function can be applied to each snippet before they are averaged in the postprocess_func
extra_sum_funcs={'domain_score': extra_sum_func}) # If additional values produced by postprocess_func need to be saved,
# it can be done using the extra_sum_funcs dictionary, which defines how to combine them.
INFO:coolpuppy:Rescaling with rescale_flank = 1 to 99x99 pixels
INFO:coolpuppy:('chr2_p', 'chr2_p'): 238
INFO:coolpuppy:('chr2_q', 'chr2_q'): 412
INFO:coolpuppy:('chr17_p', 'chr17_p'): 75
INFO:coolpuppy:('chr17_q', 'chr17_q'): 213
INFO:coolpuppy:Total number of piled up windows: 238
This is the pileup that we got from the previous step:
plotpup.plot(pup,
score=False,
height=5)
<seaborn.axisgrid.FacetGrid at 0x7ff35843abb0>

And here are the domain scores for the first 10 TADs that went into the analysis!
pup.loc[0, 'domain_score'][:10]
[1.3514487857326527,
1.0014944851906267,
1.101405324789513,
0.8571123334522476,
2.843350393430375,
1.9848390933637012,
0.7560571349817319,
1.1659901426919959,
0.9223474219342431,
1.0465908705072648]
Their distribution as a histogram:
plt.hist(pup.loc[0, 'domain_score'], bins='auto');

coolpup.py CLI¶
Use coolpup.py command to perform pileups, and plotpup.py to visualize them.
Submodules¶
coolpup.py command¶
usage: coolpup.py [-h] [--features_format {bed,bedpe,auto}] [--view VIEW]
[--flank FLANK] [--minshift MINSHIFT] [--maxshift MAXSHIFT]
[--nshifts NSHIFTS] [--expected EXPECTED] [--not_ooe]
[--mindist MINDIST] [--maxdist MAXDIST]
[--ignore_diags IGNORE_DIAGS] [--subset SUBSET]
[--by_window] [--by_strand]
[--by_distance [BY_DISTANCE [BY_DISTANCE ...]]]
[--groupby [GROUPBY [GROUPBY ...]]] [--flip_negative_strand]
[--local] [--coverage_norm [COVERAGE_NORM]] [--trans]
[--store_stripes] [--rescale]
[--rescale_flank RESCALE_FLANK]
[--rescale_size RESCALE_SIZE]
[--clr_weight_name [CLR_WEIGHT_NAME]] [-o OUTNAME]
[-p N_PROC] [--seed SEED]
[-l {DEBUG,INFO,WARNING,ERROR,CRITICAL}] [--post_mortem]
[-v]
cool_path features
Positional Arguments¶
- cool_path
Cooler file with your Hi-C data
- features
- A 3-column bed file or a 6-column double-bed file
i.e. chr1,start1,end1,chr2,start2,end2. Should be tab-delimited.
With a bed file, will consider all combinations of intervals. To pileup features along the diagonal instead, use the
--localargument.Can be piped in via stdin, then use “-”
Named Arguments¶
- --features_format, --features-format, --format, --basetype
Possible choices: bed, bedpe, auto
- Format of the features.
Options: bed: chrom, start, end bedpe: chrom1, start1, end1, chrom2, start2, end2 auto (default): determined from the file name extension Has to be explicitly provided is features is piped through stdin
Default: “auto”
- --view
Path to a file which defines which regions of the chromosomes to use
- --flank, --pad
- Flanking of the windows around the centres of specified features
i.e. final size of the matrix is 2 × flank+res, in bp. Ignored with
--rescale, use--rescale_flankinstead
Default: 100000
- --minshift
Shortest shift for random controls, bp
Default: 100000
- --maxshift
Longest shift for random controls, bp
Default: 1000000
- --nshifts
Number of control regions per averaged window
Default: 10
- --expected
- File with expected (output of
cooltools compute-expected). If None, don’t use expected and use randomly shifted controls
- File with expected (output of
- --not_ooe, --not-ooe
If expected is provided, will accumulate all expected snippets just like for randomly shifted controls, instead of normalizing each snippet individually
Default: True
- --mindist
- Minimal distance of interactions to use, bp.
If not provided, uses 2*flank+2 (in bins) as mindist to avoid first two diagonals
- --maxdist
Maximal distance of interactions to use
- --ignore_diags, --ignore-diags
How many diagonals to ignore
Default: 2
- --subset
- Take a random sample of the bed file.
Useful for files with too many featuers to run as is, i.e. some repetitive elements. Set to 0 or lower to keep all data
Default: 0
- --by_window, --by-window
- Perform by-window pile-ups.
Create a pile-up for each coordinate in the features. Not compatible with –by_strand and –by_distance.
Only works with bed format features, and generates pairwise combinations of each feature against the rest.
Default: False
- --by_strand, --by-strand
- Perform by-strand pile-ups.
Create a separate pile-up for each strand combination in the features.
Default: False
- --by_distance, --by-distance
- Perform by-distance pile-ups.
Create a separate pile-up for each distance band. If empty, will use default (0,50000,100000,200000,…) edges. Specify edges using multiple argument values, e.g. –by_distance 1000000 2000000
- --groupby
- Additional columns of features to use for groupby, space separated.
If feature_format==’bed’, each columns should be specified twice with suffixes ‘1’ and ‘2’, i.e. if features have a column ‘group’, specify ‘group1 group2’., e.g. –groupby chrom1 chrom2
- --flip_negative_strand, --flip-negative-strand
- Flip snippets so the positive strand always points to bottom-right.
Requires strands to be annotated for each feature (or two strands for bedpe format features)
Default: False
- --local
Create local pileups, i.e. along the diagonal
Default: False
- --coverage_norm, --coverage-norm
Normalize the final pileup by accumulated coverage as an alternative to balancing. Useful for single-cell Hi-C data. Can be a string: “cis” or “total” to use “cov_cis_raw” or “cov_tot_raw” columns in the cooler bin table, respectively. If they are not present, will calculate coverage with same ignore_diags as used in coolpup.py and store result in the cooler. Alternatively, if a different string is provided, will attempt to use a column with the that name in the cooler bin table, and will raise a ValueError if it does not exist. If no argument is given following the option string, will use “total”. Only allowed when using empty –clr_weight_name
Default: “”
- --trans
- Perform inter-chromosomal (trans) pileups.
This ignores all contacts in cis.
Default: False
- --store_stripes
Store horizontal and vertical stripes in pileup output
Default: False
- --rescale
- Rescale all features to the same size.
Do not use centres of features and flank, and rather use the actual feature sizes and rescale pileups to the same shape and size
Default: False
- --rescale_flank, --rescale_pad, --rescale-flank, --rescale-pad
If –rescale, flanking in fraction of feature length
Default: 1.0
- --rescale_size, --rescale-size
- Size to rescale to.
If
--rescale, used to determine the final size of the pileup, i.e. it will be size×size. Due to technical limitation in the current implementation, has to be an odd number
Default: 99
- --clr_weight_name, --weight_name, --clr-weight-name, --weight-name
- Name of the norm to use for getting balanced data.
Provide empty argument to calculate pileups on raw data (no masking bad pixels).
Default: “weight”
- -o, --outname, --output
- Name of the output file.
If not set, file is saved in the current directory and the name is generated automatically to include important information and avoid overwriting files generated with different settings.
Default: “auto”
- -p, --nproc, --n_proc, --n-proc
- Number of processes to use.
Each process works on a separate chromosome, so might require quite a bit more memory, although the data are always stored as sparse matrices. Set to 0 to use all available cores.
Default: 1
- --seed
Set specific seed value to ensure reproducibility
- -l, --log
Possible choices: DEBUG, INFO, WARNING, ERROR, CRITICAL
Set the logging level
Default: “INFO”
- --post_mortem, --post-mortem
Enter debugger if there is an error
Default: False
- -v, --version
show program’s version number and exit
dividepups.py command¶
usage: dividepups.py [-h] [-v] [-o OUTNAME] input_pups [input_pups ...]
Positional Arguments¶
- input_pups
Two pileups to divide
Named Arguments¶
- -v, --version
show program’s version number and exit
- -o, --outname
- Name of the output file.
If not set, file is saved in the current directory and the name is generated automatically.
Default: “auto”
plotpup.py command¶
usage: plotpup.py [-h] [--cmap CMAP] [--not_symmetric] [--vmin VMIN]
[--vmax VMAX] [--scale {linear,log}] [--stripe STRIPE]
[--stripe_sort STRIPE_SORT] [--lineplot]
[--out_sorted_bedpe OUT_SORTED_BEDPE] [--divide_pups]
[--font FONT] [--font_scale FONT_SCALE] [--cols COLS]
[--rows ROWS] [--col_order COL_ORDER]
[--row_order ROW_ORDER] [--colnames COLNAMES [COLNAMES ...]]
[--rownames ROWNAMES [ROWNAMES ...]] [--query QUERY]
[--norm_corners NORM_CORNERS] [--no_score] [--center CENTER]
[--ignore_central IGNORE_CENTRAL] [--quaich] [--dpi DPI]
[--height HEIGHT] [--plot_ticks] [--output OUTPUT]
[-l {DEBUG,INFO,WARNING,ERROR,CRITICAL}] [--post_mortem]
[--input_pups INPUT_PUPS [INPUT_PUPS ...]] [-v]
Named Arguments¶
- --cmap
- Colormap to use
Default: “coolwarm”
- --not_symmetric, --not-symmetric, --not_symmetrical, --not-symmetrical
Whether to not make colormap symmetric around 1, if log scale
Default: False
- --vmin
Value for the lowest colour
- --vmax
Value for the highest colour
- --scale
Possible choices: linear, log
Whether to use linear or log scaling for mapping colours
Default: “log”
- --stripe
For plotting stripe stackups
- --stripe_sort
Whether to sort stripe stackups by total signal (sum), central pixel signal (center_pixel), or not at all (None)
Default: “sum”
- --lineplot
- Whether to plot the average lineplot above stripes.
This only works for a single plot, i.e. without rows/columns
Default: False
- --out_sorted_bedpe
Output bedpe of sorted stripe regions
- --divide_pups
Whether to divide two pileups and plot the result
Default: False
- --font
Font to use for plotting
Default: “DejaVu Sans”
- --font_scale
Font scale to use for plotting. Defaults to 1
Default: 1
- --cols
Which value to map as columns
- --rows
Which value to map as rows
- --col_order
Order of columns to use, space separated inside quotes
- --row_order
Order of rows to use, space separated inside quotes
- --colnames
Names to plot for columns, space separated.
- --rownames
Names to plot for rows, space separated.
- --query
- Pandas query to select pups to plot from concatenated input files.
Multiple query arguments can be used. Usage example: –query “orientation == ‘+-’ | orientation == ‘-+’”
- --norm_corners
- Whether to normalize pileups by their top left and bottom right corners.
0 for no normalization, positive number to define the size of the corner squares whose values are averaged
Default: 0
- --no_score
If central pixel score should not be shown in top left corner
Default: False
- --center
How many central pixels to consider when calculating enrichment for off-diagonal pileups.
Default: 3
- --ignore_central
How many central bins to ignore when calculating insulation for local (on-diagonal) non-rescaled pileups.
Default: 3
- --quaich
Activate if pileups are named accodring to Quaich naming convention to get information from the file name
Default: False
- --dpi
DPI of the output plot. Try increasing if heatmaps look blurry
Default: 300
- --height
Height of the plot
Default: 1
- --plot_ticks
Whether to plot ticks demarkating the center and flanking regions, only applicable for non-stripes
Default: False
- --output, -o, --outname
Where to save the plot
Default: “pup.pdf”
- -l, --log
Possible choices: DEBUG, INFO, WARNING, ERROR, CRITICAL
Set the logging level
Default: “INFO”
- --post_mortem
Enter debugger if there is an error
Default: False
- --input_pups
All files to plot
- -v, --version
show program’s version number and exit
coolpuppy Python API¶
While coolpup.py was designed with CLI in mind, it’s possible to use the classes and functions directly in Python code to perform pileups.
coolpuppy.coolpup module¶
- class coolpuppy.coolpup.CoordCreator(features, resolution, *, features_format='auto', flank=100000, rescale_flank=None, chroms='all', minshift=100000, maxshift=1000000, nshifts=10, mindist='auto', maxdist=None, local=False, subset=0, trans=False, seed=None)¶
Bases:
object- __init__(features, resolution, *, features_format='auto', flank=100000, rescale_flank=None, chroms='all', minshift=100000, maxshift=1000000, nshifts=10, mindist='auto', maxdist=None, local=False, subset=0, trans=False, seed=None)¶
Generator of coordinate pairs for pileups.
- Parameters
features (DataFrame) – A bed- or bedpe-style file with coordinates.
resolution (int, optional) – Data resolution.
features_format (str, optional) –
- Format of the features. Options:
bed: chrom, start, end bedpe: chrom1, start1, end1, chrom2, start2, end2 auto (default): determined from the columns in the DataFrame
flank (int, optional) – Padding around the central bin, in bp. For example, with 5000 bp resolution and 100000 flank, final pileup is 205000×205000 bp. The default is 100000.
rescale_flank (float, optional) – Fraction of ROI size added on each end when extracting snippets, if rescale. The default is None. If specified, overrides flank.
chroms (str or list, optional) – Which chromosomes to use for pileups. Has to be in a list even for a single chromosome, e.g. [‘chr1’]. The default is “all”
minshift (int, optional) – Minimal shift applied when generating random controls, in bp. The default is 10 ** 5.
maxshift (int, optional) – Maximal shift applied when generating random controls, in bp. The default is 10 ** 6.
nshifts (int, optional) – How many shifts to generate per region of interest. Does not take chromosome boundaries into account The default is 10.
mindist (int, optional) – Shortest interactions to consider. Uses midpoints of regions of interest. “auto” selects it to avoid the two shortest diagonals of the matrix, i.e. 2 * flank + 2 * resolution The default is “auto”.
maxdist (int, optional) – Longest interactions to consider. The default is None.
local (bool, optional) – Whether to generate local coordinates, i.e. on-diagonal. The default is False.
subset (int, optional) – What subset of the coordinate files to use. 0 or negative to use all. The default is 0.
seed (int, optional) – Seed for np.random to make it reproducible. The default is None.
trans (bool, optional) – Whether to generate inter-chromosomal (trans) pileups. The default is False
- Return type
Object that generates coordinates for pileups required for PileUpper.
- bedpe2bed(df, ends=True, how='center')¶
- empty_stream(*args, **kwargs)¶
- filter_func_all(intervals)¶
- filter_func_chrom(chrom)¶
- filter_func_region(region)¶
- filter_func_trans_pairs(region1, region2)¶
- get_combinations(filter_func1, filter_func2=None, intervals=None, control=False, groupby=[], modify_2Dintervals_func=None)¶
- get_intervals_stream(filter_func1, filter_func2=None, intervals=None, control=False, groupby=[], modify_2Dintervals_func=None)¶
- process()¶
- class coolpuppy.coolpup.PileUpper(clr, CC, *, view_df=None, clr_weight_name='weight', expected=False, expected_value_col='balanced.avg', ooe=True, control=False, coverage_norm=False, rescale=False, rescale_size=99, flip_negative_strand=False, ignore_diags=2, store_stripes=False, nproc=1)¶
Bases:
object- __init__(clr, CC, *, view_df=None, clr_weight_name='weight', expected=False, expected_value_col='balanced.avg', ooe=True, control=False, coverage_norm=False, rescale=False, rescale_size=99, flip_negative_strand=False, ignore_diags=2, store_stripes=False, nproc=1)¶
Creates pileups
- Parameters
clr (cool) – Cool file with Hi-C data.
CC (CoordCreator) – CoordCreator object with correct settings.
clr_weight_name (bool or str, optional) – Whether to use balanced data, and which column to use as weights. The default is “weight”. Provide False to use raw data.
expected (DataFrame, optional) – If using expected, pandas DataFrame with by-distance expected. The default is False.
view_df (DataFrame) – A dataframe with region coordinates used in expected (see bioframe documentation for details). Can be ommited if no expected is provided, or expected is for whole chromosomes.
ooe (bool, optional) – Whether to normalize each snip by expected value. If False, all snips are accumulated, all expected values are accumulated, and then the former divided by the latter - like with randomly shifted controls. Only has effect when expected is provided.
control (bool, optional) – Whether to use randomly shifted controls. The default is False.
coverage_norm (bool or str, optional) – Whether to normalize final the final pileup by accumulated coverage as an alternative to balancing. Useful for single-cell Hi-C data. Can be either boolean, or string: “cis” or “total” to use “cov_cis_raw” or “cov_tot_raw” columns in the cooler bin table, respectively. If True, will attempt to use “cov_tot_raw” if available, otherwise will compute and store coverage in the cooler with default column names, and use “cov_tot_raw”. Alternatively, if a different string is provided, will attempt to use a column with the that name in the cooler bin table, and will raise a ValueError if it does not exist. Only allowed when clr_weight_name is False. The default is False.
rescale (bool, optional) – Whether to rescale the pileups. The default is False
rescale_size (int, optional) – Final shape of rescaled pileups. E.g. if 99, pileups will be squares of 99×99 pixels. The default is 99.
flip_negative_strand (bool, optional) – Flip snippets so the positive strand always points to bottom-right. Requires strands to be annotated for each feature (or two strands for bedpe format features)
ignore_diags (int, optional) – How many diagonals to ignore to avoid short-distance artefacts. The default is 2.
store_stripes (bool, optional) – Whether to store horizontal and vertical stripes and coordinates in the output The default is False
nproc (int, optional) – Number of processes to use. The default is 1.
- Return type
Object that generates pileups.
- accumulate_stream(snip_stream, postprocess_func=None, extra_funcs=None)¶
- Parameters
snip_stream (generator) –
- Generator of pd.Series, each one containing at least:
a snippet as a 2D array in [‘data’], [‘cov_start’] and [‘cov_end’] as 1D arrays (can be all 0)
And any other annotations
postprocess_func (function, optional) – Any additional postprocessing of each snip needed, in one function. Can be used to modify the data in un-standard way, or create groups when it can’t be done before snipping, or to assign each snippet to multiple groups. Example: lib.puputils.group_by_region.
extra_funcs (dict, optional) – Any additional functions to be applied every time a snip is added to a pileup or two pileups are summed up - see _add_snip and sum_pups.
- Returns
outdict – Dictionary of accumulated snips (each as a Series) for each group. Always includes “all”
- Return type
dict
- get_data(region1, region2=None)¶
Get sparse data for a region
- Parameters
region1 (tuple or str) – Region for which to load the data. Either tuple of (chr, start, end), or string with region name.
region2 (tuple or str, optional) – Second region for between which and the first region to load the data. Either tuple of (chr, start, end), or string with region name. Default is None
- Returns
data – Sparse csr matrix for the corresponding region.
- Return type
csr
- get_expected_trans(region1, region2)¶
- make_outmap()¶
Generate zero-filled array of the right shape
- Returns
outmap – Array of zeros of correct shape.
- Return type
array
- pileup_region(region1, region2=None, groupby=[], modify_2Dintervals_func=None, postprocess_func=None, extra_sum_funcs=None)¶
- Parameters
region1 (str) – Region name.
region2 (str, optional) – Region name.
groupby (list of str, optional) – Which attributes of each snip to assign a group to it
modify_2Dintervals_func (function, optional) – A function to apply to a dataframe of genomic intervals used for pileups. If possible, much preferable to postprocess_func for better speed. Good example is the bin_distance_intervals function above.
postprocess_func (function, optional) – Additional function to apply to each snippet before grouping. Good example is the lib.puputils.bin_distance function, but using bin_distance_intervals as modify_2Dintervals_func is much faster.
extra_sum_funcs (dict, optional) – Any additional functions to be applied every time a snip is added to a pileup or two pileups are summed up - see _add_snip and sum_pups.
- Returns
pileup – accumulated snips as a dict
- Return type
dict
- pileupsByDistanceWithControl(nproc=None, distance_edges='default', groupby=[])¶
Perform by-distance pileups across all chromosomes and applies required normalization. Simple wrapper around pileupsWithControl
- Parameters
nproc (int, optional) – How many cores to use. Sends a whole chromosome per process. The default is None, which uses the same number as nproc set at creation of the object.
distance_edges (list/array of int) – How to group snips by distance (based on their centres). Default uses separations [0, 50_000, 100_000, 200_000, …]
groupby (list of str, optional) – Which attributes of each snip to assign a group to it
- Returns
pileup_df – Normalized pileups in a pandas DataFrame, with columns data and num. data contains the normalized pileups, and num - how many snippets were combined (the regions of interest, not control regions). Each distance band is a row, annotated in column distance_band
- Return type
2D array
- pileupsByStrandByDistanceWithControl(nproc=None, distance_edges='default', groupby=[])¶
Perform by-strand by-distance pileups across all chromosomes and applies required normalization. Simple wrapper around pileupsWithControl. Assumes the features in CoordCreator file has a “strand” column.
- Parameters
nproc (int, optional) – How many cores to use. Sends a whole chromosome per process. The default is None, which uses the same number as nproc set at creation of the object.
distance_edges (list/array of int) – How to group snips by distance (based on their centres). Default uses separations [0, 50_000, 100_000, 200_000, …]
groupby (list of str, optional) – Which attributes of each snip to assign a group to it
- Returns
pileup_df – Normalized pileups in a pandas DataFrame, with columns data and num. data contains the normalized pileups, and num - how many snippets were combined (the regions of interest, not control regions). Each distance band is a row, annotated in columns separation
- Return type
2D array
- pileupsByStrandWithControl(nproc=None, groupby=[])¶
Perform by-strand pileups across all chromosomes and applies required normalization. Simple wrapper around pileupsWithControl. Assumes the features in CoordCreator file has a “strand” column.
- Parameters
nproc (int, optional) – How many cores to use. Sends a whole chromosome per process. The default is None, which uses the same number as nproc set at creation of the object.
groupby (list of str, optional) – Which attributes of each snip to assign a group to it
- Returns
pileup_df – Normalized pileups in a pandas DataFrame, with columns data and num. data contains the normalized pileups, and num - how many snippets were combined (the regions of interest, not control regions). Each distance band is a row, annotated in columns separation
- Return type
2D array
- pileupsByWindowWithControl(nproc=None)¶
Perform by-window (i.e. for each region) pileups across all chromosomes and applies required normalization. Simple wrapper around pileupsWithControl
- Parameters
nproc (int, optional) – How many cores to use. Sends a whole chromosome per process. The default is None, which uses the same number as nproc set at creation of the object.
- Returns
pileup_df – Normalized pileups in a pandas DataFrame, with columns data and num. data contains the normalized pileups, and num - how many snippets were combined (the regions of interest, not control regions). Each window is a row (coordinates are recorded in columns [‘chrom’, ‘start’, ‘end’]), plus an additional row is created with all data (with “all” in the “chrom” column and -1 in start and end).
- Return type
2D array
- pileupsWithControl(nproc=None, groupby=[], modify_2Dintervals_func=None, postprocess_func=None, extra_sum_funcs=None)¶
Perform pileups across all chromosomes and applies required normalization
- Parameters
nproc (int, optional) – How many cores to use. Sends a whole chromosome per process. The default is None, which uses the same number as nproc set at creation of the object.
groupby (list of str, optional) – Which attributes of each snip to assign a group to it
modify_2Dintervals_func (function, optional) – Function to apply to the DataFrames of coordinates before fetching snippets based on them. Preferable to using the postprocess_func, since at the earlier stage it can be vectorized and much more efficient.
postprocess_func (function, optional) – Additional function to apply to each snippet before grouping. Good example is the lib.puputils.bin_distance function.
extra_sum_funcs (dict, optional) – Any additional functions to be applied every time a snip is added to a pileup or two pileups are summed up - see _add_snip and sum_pups.
- Returns
pileup_df – Normalized pileups in a pandas DataFrame, with columns data and num. data contains the normalized pileups, and num - how many snippets were combined (the regions of interest, not control regions). Each condition from groupby is a row, plus an additional row all is created with all data.
- Return type
2D array
- coolpuppy.coolpup.assign_groups(intervals, groupby=[])¶
Assign groups to rows based on a list of columns
- Parameters
intervals (pd.DataFrame) – Dataframe containing intervals with any annotations.
groupby (list, optional) – List of columns to use to assign a group. The default is [].
- Returns
intervals – Adds a “group” column with the annotation based on groupby. If groupby is empty, assigns “all” to all rows.
- Return type
pd.DataFrame
- coolpuppy.coolpup.bin_distance_intervals(intervals, band_edges='default')¶
- Parameters
intervals (pd.DataFrame) – Dataframe containing intervals with any annotations. Has to have a ‘distance’ column
band_edges (list or array-like, or "default", optional) – Edges of distance bands used to split the intervals into groups. Default is np.append([0], 50000 * 2 ** np.arange(30))
- Returns
snip – The same dataframe with added [‘distance_band’] annotation.
- Return type
pd.DataFrame
- coolpuppy.coolpup.expand(intervals, flank, resolution, rescale_flank=None)¶
- coolpuppy.coolpup.expand2D(intervals, flank, resolution, rescale_flank=None)¶
- coolpuppy.coolpup.pileup(clr, features, features_format='bed', view_df=None, expected_df=None, expected_value_col='balanced.avg', clr_weight_name='weight', flank=100000, minshift=100000, maxshift=1000000, nshifts=0, ooe=True, mindist='auto', maxdist=None, min_diag=2, subset=0, by_window=False, by_strand=False, by_distance=False, groupby=[], flip_negative_strand=False, local=False, coverage_norm=False, trans=False, rescale=False, rescale_flank=1, rescale_size=99, store_stripes=False, nproc=1, seed=None)¶
Create pileups
- Parameters
clr (cool) – Cool file with Hi-C data.
features (DataFrame) – A bed- or bedpe-style file with coordinates.
features_format (str, optional) –
- Format of the features. Options:
bed: chrom, start, end bedpe: chrom1, start1, end1, chrom2, start2, end2 auto (default): determined from the columns in the DataFrame
view_df (DataFrame) – A dataframe with region coordinates used in expected (see bioframe documentation for details). Can be ommited if no expected is provided, or expected is for whole chromosomes.
expected_df (DataFrame, optional) – If using expected, pandas DataFrame with by-distance expected. The default is False.
expected_value_col (str, optional) – Which column in the expected_df contains values to use for normalization
clr_weight_name (bool or str, optional) – Whether to use balanced data, and which column to use as weights. The default is “weight”. Provide False to use raw data.
flank (int, optional) – Padding around the central bin, in bp. For example, with 5000 bp resolution and 100000 flank, final pileup is 205000×205000 bp. The default is 100000.
minshift (int, optional) – Minimal shift applied when generating random controls, in bp. The default is 10 ** 5.
maxshift (int, optional) – Maximal shift applied when generating random controls, in bp. The default is 10 ** 6.
nshifts (int, optional) – How many shifts to generate per region of interest. Does not take chromosome boundaries into account The default is 10.
ooe (bool, optional) – Whether to normalize each snip by expected value. If False, all snips are accumulated, all expected values are accumulated, and then the former divided by the latter - like with randomly shifted controls. Only has effect when expected is provided. Default is True.
mindist (int, optional) – Shortest interactions to consider. Uses midpoints of regions of interest. “auto” selects it to avoid the two shortest diagonals of the matrix, i.e. 2 * flank + 2 * resolution The default is “auto”.
maxdist (int, optional) – Longest interactions to consider. The default is None.
min_diag (int, optional) – How many diagonals to ignore to avoid short-distance artefacts. The default is 2.
subset (int, optional) – What subset of the coordinate files to use. 0 or negative to use all. The default is 0.
by_window (bool, optional) – Whether to create a separate pileup for each feature by accumulating all of its interactions with other features. Produces as many pileups, as there are features. The default is False.
by_strand (bool, optional) – Whether to create a separate pileup for each combination of “strand1”, “strand2” in features. If features_format==’bed’, first creates pairwise combinations of features, and the original features need to have a column “strand”. If features_format==’bedpe’, they need to have “strand1” and “strand2” columns. The default is False.
by_distance (bool or list, optional) –
Whether to create a separate pileup for different distance separations. If features_format==’bed’, internally creates pairwise combinations of features. If True, splits all separations using edges defined like this:
band_edges = np.append([0], 50000 * 2 ** np.arange(30))
Alternatively, a list of integer values can be given with custom distance edges. The default is False.
groupby (list of str, optional) – Additional columns of features to use for groupby. If feature_format==’bed’, each columns should be specified twice with suffixes “1” and “2”, i.e. if features have a column “group”, specify [“group1”, “group2”]. The default is [].
flip_negative_strand (bool, optional) – Flip snippets so the positive strand always points to bottom-right. Requires strands to be annotated for each feature (or two strands for bedpe format features)
local (bool, optional) – Whether to generate local coordinates, i.e. on-diagonal. The default is False.
coverage_norm (bool or str, optional) – Whether to normalize final the final pileup by accumulated coverage as an alternative to balancing. Useful for single-cell Hi-C data. Can be either boolean, or string: “cis” or “total” to use “cov_cis_raw” or “cov_tot_raw” columns in the cooler bin table, respectively. If True, will attempt to use “cov_tot_raw” if available, otherwise will compute and store coverage in the cooler with default column names, and use “cov_tot_raw”. Alternatively, if a different string is provided, will attempt to use a column with the that name in the cooler bin table, and will raise a ValueError if it does not exist. Only allowed when clr_weight_name is False. The default is False.
trans (bool, optional) – Whether to generate inter-chromosomal (trans) pileups. The default is False
rescale (bool, optional) – Whether to rescale the pileups. The default is False
rescale_flank (float, optional) – Fraction of ROI size added on each end when extracting snippets, if rescale. The default is None. If specified, overrides flank.
rescale_size (int, optional) – Final shape of rescaled pileups. E.g. if 99, pileups will be squares of 99×99 pixels. The default is 99.
store_stripes (bool, optional) – Whether to store horizontal and vertical stripes and coordinates in the output The default is False
nproc (int, optional) – Number of processes to use. The default is 1.
seed (int, optional) – Seed for np.random to make it reproducible. The default is None.
- Returns
pileup_df - pandas DataFrame containing the pileups and their grouping information,
if any, all possible annotations from the arguments of this function.
coolpuppy.lib.io module¶
- coolpuppy.lib.io.is_gz_file(filepath)¶
- coolpuppy.lib.io.load_array_with_header(filename)¶
Load array from files generated using save_array_with_header. They are simple txt files with an optional header in the first lines, commented using “# “. If uncommented, the header is in YAML.
- Parameters
filename (string) – File to load from.
- Returns
data – Dictionary with information from the header. Access the associated data in an array using data[‘data’].
- Return type
dict
- coolpuppy.lib.io.load_pileup_df(filename, quaich=False, skipstripes=False)¶
Loads a dataframe saved using save_pileup_df
- Parameters
filename (str) – File to load from.
quaich (bool, optional) – Whether to assume standard quaich file naming to extract sample name and bedname. The default is False.
- Returns
annotation – Pileups are in the “data” column, all metadata in other columns
- Return type
pd.DataFrame
- coolpuppy.lib.io.load_pileup_df_list(files, quaich=False, nice_metadata=True, skipstripes=False)¶
- Parameters
files (iterable) – Files to read pileups from.
quaich (bool, optional) – Whether to assume standard quaich file naming to extract sample name and bedname. The default is False.
nice_metadata (bool, optional) – Whether to add nicer metadata for direct plotting. The default is True. Adds a “norm” column (“expected”, “shifts” or “none”).
- Returns
pups – Combined dataframe with all pileups and annotations from all files.
- Return type
pd.DataFrame
- coolpuppy.lib.io.save_array_with_header(array, header, filename)¶
Save a numpy array with a YAML header generated from a dictionary
- Parameters
array (np.array) – Array to save.
header (dict) – Dictionaty to save into the header.
filename (string) – Name of file to save array and metadata into.
- coolpuppy.lib.io.save_pileup_df(filename, df, metadata=None, mode='w', compression='lzf')¶
Saves a dataframe with metadata into a binary HDF5 file`
- Parameters
filename (str) – File to save to.
df (pd.DataFrame) – DataFrame to save into binary hdf5 file.
metadata (dict, optional) – Dictionary with meatadata.
mode (str, optional) – Mode for the first time access to the output file: ‘w’ to overwrite if file exists, or ‘a’ to fail if output file already exists
compression (str, optional) – Compression to use for saving, e.g. ‘gzip’. Defaults to ‘lzf’
- Return type
None.
Notes
Replaces None in metadata values with False, since HDF5 doesn’t support None
- coolpuppy.lib.io.sniff_for_header(file, sep='\t', comment='#')¶
Warning: reads the entire file into a StringIO buffer!
coolpuppy.lib.numutils module¶
- coolpuppy.lib.numutils.corner_cv(amap, i=4)¶
Get coefficient of variation for upper left and lower right corners of a pileup to estimate how noisy it is
- Parameters
amap (2D array) – Pileup.
i (int, optional) – How many bins to use from each upper left and lower right corner: final corner shape is i^2. The default is 4.
- Returns
CV – Coefficient of variation for the corner pixels.
- Return type
float
- coolpuppy.lib.numutils.get_domain_score(amap, flank=1)¶
Divide sum of values in a square from the central part of a matrix by the upper and right rectangles corresponding to interactions of the central region with its surroundings.
- Parameters
amap (2D array) – Pileup.
flank (int) – Relative padding used, i.e. if 1 the central third is used, if 2 the central fifth is used. The default is 1.
- Returns
score – Domain score.
- Return type
float
- coolpuppy.lib.numutils.get_enrichment(amap, n)¶
Get values from the center of a pileup for a square with side n
- Parameters
amap (2D array) – Pileup.
n (int) – Side of the central square to use.
- Returns
enrichment – Mean of the pixels in the central square.
- Return type
float
- coolpuppy.lib.numutils.get_insulation_strength(amap, ignore_central=0, ignore_diags=2)¶
Divide values in upper left and lower right corners over upper right and lower left, ignoring the central bins.
- Parameters
amap (2D array) – Pileup.
ignore_central (int, optional) – How many central bins to ignore. Has to be odd or 0. The default is 0.
- Returns
Insulation strength.
- Return type
float
- coolpuppy.lib.numutils.get_local_enrichment(amap, flank=1)¶
Get values for a square from the central part of a pileup, ignoring padding
- Parameters
amap (2D array) – Pileup.
flank (int) – Relative padding used, i.e. if 1 the central third is used, if 2 the central fifth is used. The default is 1.
- Returns
enrichment – Mean of the pixels in the central square.
- Return type
float
- coolpuppy.lib.numutils.norm_cis(amap, i=3)¶
Normalize the pileup by mean of pixels from upper left and lower right corners
- Parameters
amap (2D array) – Pileup.
i (int, optional) – How many bins to use from each upper left and lower right corner: final corner shape is i^2. 0 will not normalize. The default is 3.
- Returns
amap – Normalized pileup.
- Return type
2D array
coolpuppy.lib.puputils module¶
- coolpuppy.lib.puputils.accumulate_values(dict1, dict2, key)¶
Useful as an extra_sum_func
- coolpuppy.lib.puputils.bin_distance(snip, band_edges='default')¶
- Parameters
snip (pd.Series) – Series containing any annotations. Has to have [‘distance’]
band_edges (list or array-like, or "default", optional) – Edges of distance bands used to assign the distance band. Default is np.append([0], 50000 * 2 ** np.arange(30))
- Returns
snip – The same snip with added [‘distance_band’] annotation.
- Return type
pd.Series
- coolpuppy.lib.puputils.divide_pups(pup1, pup2)¶
Divide two pups and get the resulting pup. Requires that the pups have identical shapes, resolutions, flanks, etc. If pups contain stripes, these will only be divided if stripes have identical coordinates.
- coolpuppy.lib.puputils.get_score(pup, center=3, ignore_central=3)¶
Calculate a reasonable score for any kind of pileup For non-local (off-diagonal) pileups, calculates average signal in the central pixels (based on ‘center’). For local non-rescaled pileups calculates insulation strength, and ignores the central bins (based on ‘ignore_central’) For local rescaled pileups calculates enrichment in the central rescaled area relative to the two neighouring areas on the sides.
- Parameters
pup (pd.Series or dict) – Series or dict with pileup in ‘data’ and annotations in other keys. Will correctly calculate enrichment score with annotations in ‘local’ (book), ‘rescale’ (bool) and ‘rescale_flank’ (float)
enrichment (int, optional) – Passed to ‘get_enrichment’ to calculate the average strength of central pixels. The default is 3.
ignore_central (int, optional) – How many central bins to ignore for calculation of insulation in local pileups. The default is 3.
- Returns
Score.
- Return type
float
- coolpuppy.lib.puputils.group_by_region(snip)¶
- coolpuppy.lib.puputils.norm_coverage(snip)¶
Normalize a pileup by coverage arrays
- Parameters
loop (2D array) – Pileup.
cov_start (1D array) – Accumulated coverage of the left side of the pileup.
cov_end (1D array) – Accumulated coverage of the bottom side of the pileup.
- Returns
loop – Normalized pileup.
- Return type
2D array
- coolpuppy.lib.puputils.sum_pups(pup1, pup2, extra_funcs={})¶
Preserves data, stripes, cov_start, cov_end, n, num and coordinates Assumes n=1 if not present, and calculates num if not present If store_stripes is set to False, stripes and coordinates will be empty
extra_funcs allows to give arbitrary functions to accumulate extra information from the two pups.
coolpuppy.plotpup module¶
- coolpuppy.plotpup.add_heatmap(data, flank, rescale, rescale_flank, n, max_coordinates, height=1, aspect='auto', color=None, cmap='coolwarm', norm=<Mock name='mock.LogNorm()' id='139859660105744'>, plot_ticks=False, stripe=False, font_scale=1)¶
Adds the array contained in data.values[0] to the current axes as a heatmap of stripes
- coolpuppy.plotpup.add_score(score, height=1, color=None, font_scale=1)¶
Adds the value contained in score.values[0] to the current axes as a label in top left corner
- coolpuppy.plotpup.add_stripe_lineplot(data, resolution, flank, rescale, rescale_flank, height=1, aspect='auto', color=None, cmap='coolwarm', scale='log', norm=<Mock name='mock.LogNorm()' id='139859660105744'>, plot_ticks=False, stripe=False, font_scale=1, colnames=None)¶
Adds the array contained in data.values[0] to the current axes as a heatmap of stripes and an average lineplot on top. Only works with one condition at a time.
- coolpuppy.plotpup.auto_rows_cols(n)¶
Automatically determines number of rows and cols for n pileups
- Parameters
n (int) – Number of pileups.
- Returns
rows (int) – How many rows to use.
cols (int) – How many columsn to use.
- coolpuppy.plotpup.get_min_max(pups, vmin=None, vmax=None, sym=True, scale='log')¶
Automatically determine minimal and maximal colour intensity for pileups
- Parameters
pups (np.array) – Numpy array of numpy arrays conaining pileups.
vmin (float, optional) – Force certain minimal colour. The default is None.
vmax (float, optional) – Force certain maximal colour. The default is None.
sym (bool, optional) – Whether the output should be cymmetrical around 0. The default is True.
- Returns
vmin (float) – Selected minimal colour.
vmax (float) – Selected maximal colour.
- coolpuppy.plotpup.plot(pupsdf, cols=None, rows=None, score='score', center=3, ignore_central=3, col_order=None, row_order=None, vmin=None, vmax=None, sym=True, norm_corners=0, cmap='coolwarm', cmap_emptypixel=(0.98, 0.98, 0.98), scale='log', height=1, aspect=1, font='DejaVu Sans', font_scale=1, plot_ticks=False, colnames=None, rownames=None, **kwargs)¶
- coolpuppy.plotpup.plot_stripes(pupsdf, cols=None, rows=None, col_order=None, row_order=None, vmin=None, vmax=None, sym=True, cmap='coolwarm', cmap_emptypixel=(0.98, 0.98, 0.98), scale='log', height=1, aspect='auto', stripe='corner_stripe', stripe_sort='sum', out_sorted_bedpe=None, font='DejaVu Sans', font_scale=1, plot_ticks=False, colnames=None, rownames=None, lineplot=False, **kwargs)¶
- coolpuppy.plotpup.sort_separation(sep_string_series, sep='Mb')¶